Chapter: Modern Analytical Chemistry: Electrochemical Methods of Analysis
Quantitative Applications - Voltammetric Methods of Analysis
Quantitative Applications
Quantitative voltammetry has been applied
to a wide variety of sample
types, including environmental samples, clinical samples,
pharmaceu-tical formulations, steels, gasoline, and oil.
Selecting the Voltammetric Technique
The
choice of which
voltammetric tech- nique to
use depends on the sample’s characteristics, including the analyte’s ex- pected concentration and the location of the sample.
Amperometry is best
suited for use as a detector
in flow systems or as a selective
sensor for the rapid analysis
of a single analyte.
The portability of amperometric sensors,
which are similar
to po- tentiometric sensors,
make them ideal
for field studies.
Pulse polarography and stripping voltammetry can frequently be
used inter- changeably, although each has its advantages and disadvantages.
Pulse polarography is better for analyzing
a wider range of inorganic
and organic analytes
because the need to preconcentrate the analyte at the electrode surface restricts the application
of anodic and cathodic stripping
voltammetry.
When either pulse polarography or anodic stripping
voltammetry can be used,
the selection is often based
on the analyte’s expected concentration and the desired accuracy and precision. Detection
limits for normal pulse polarography generally are on the order of 10–6–10–7 M, whereas
those for differential pulse polarography,
staircase, and square-wave polarography are between
10–7 M
and 10–8
M. Precon- centrating the analyte in stripping voltammetry lowers the detection limit for many analytes to as little
as 10–10
M. On the
other hand, the
current in stripping voltam- metry is much more sensitive
than pulse polarography to changes in experimental
conditions, which may lead to poorer precision and accuracy.
Anodic
stripping voltammetry also suffers from occasional interferences
when
two metals, such as Cu and Zn, combine to form an intermetallic com- pound in the mercury
amalgam. The deposition potential for Zn2+ is sufficiently
negative that any Cu2+ present in the sample
is also deposited. After deposition, intermetallic compounds such as CuZn and CuZn2 form within the mercury
amalgam. During the stripping step, the zinc in the intermetallic compounds
strips at potentials near that of copper, decreasing the current for zinc and in- creasing the current for copper. This problem can often be overcome by adding a third
element that forms
a stronger intermetallic compound with the interfering
metal. Thus, adding Ga3+ minimizes this problem by forming an intermetallic
compound of Cu and Ga.
Correcting for Residual Current
In any quantitative analysis
the signal due to the analyte must be corrected for signals arising
from other sources. The total measured current in any voltammetric experiment, itot, consists of two
parts: that due
to the analyte’s oxidation or reduction, ia, and a background, or residual, current,
ir.
itot = ia +
ir
The residual current, in turn, has
two sources. One
source is a faradaic current
due to the oxidation or reduction of trace impurities in the sample,
ii. The other source is the charging current,
ich, that is present
whenever the working
electrode’s poten- tial changes.
ir = ii + ich
Faradaic currents due
to impurities can
usually be minimized by carefully preparing the sample. For example,
one important impurity
is dissolved O2, which is reduced first to H2O2 and then to H2O. Dissolved O2 is removed
by bubbling an inert gas such as N2 through
the sample before the analysis.
Two methods are commonly used to correct
for the residual current. One method is to extrapolate the total measured
current when the analyte’s faradaic current is zero.. The advantage of this method
is that it does not require any additional
data. On the other hand,
extrapolation assumes that changes in the residual
cur- rent with potential are predictable, which
often is not the case.
A second, and more rigorous, approach is to obtain a voltammogram for
an appropriate blank. The blank’s residual current
is then subtracted from the total
current obtained with the sample.
Analysis for Single Components
The analysis of
samples containing only a single electroactive analyte is straightforward. Any
of the standardization methods can be used to establish
the relationship between
current and the concentration
of analyte.


Multicomponent Analysis
One
advantage of
voltammetry as
a quantitative method of analysis is its capability for analyzing two
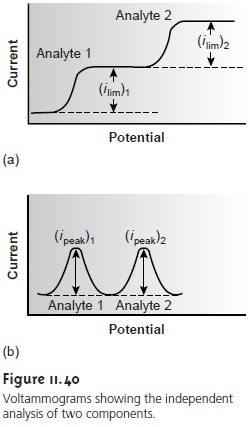
or more analytes in a single sample. As long as the components behave independently, the resulting voltammo- gram for a multicomponent
mixture is a summation of their respective individual
voltammograms. If the separation between
the half-wave potentials or peak poten- tials is sufficient, each component can be determined independently as if it were the
only component in the sample
(Figure 11.40). The minimum separation between the half-wave potentials or peak potentials for the independent analysis of two com-
ponents depends on several factors,
including the type of electrode and the poten- tial-excitation signal. For
normal polarography the
separation must be at least ±0.2–0.3 V, and for differential pulse voltammetry a
minimum separation of ±0.04–0.05 V is
needed.
When
the
overlap
between
the
voltammograms for two components pre-
vents their independent analysis, a simultaneous analysis
similar to that used in
spectrophotometry may be possible. An example of this approach
is outlined in Example 11.12.

Environmental Samples
One area in which quantitative voltammetry has had a
significant effect is in the analysis of trace metals
in environmental samples.
The most common samples
are natural waters,
including groundwater, lakes,
rivers and streams, sea water, rain,
and snow. Concentrations of trace metals
at the parts-per- billion level can be determined using differential pulse polarography, whereas
with anodic stripping voltammetry the determination of trace metals at the pptr (parts- per-trillion) level is possible.
The combination of low detection
limits and the capa-
bility for the simultaneous analysis
of several analytes
makes differential pulse
po- larography and anodic
stripping voltammetry ideally
suited for such samples.
One interesting application of anodic stripping voltammetry to the analysis of natural waters is the determination of the speciation, or chemical form,
of the trace metals. The speciation of a trace
metal is important because its bioavailability, toxi- city, and ease of transport
through the environment often depend on its chemical form. For example, trace
metals strongly bound
to colloidal particles are generally not
available to aquatic lifeforms and, therefore, are not toxic. Unfortunately, an- odic
stripping voltammetry cannot
distinguish the exact
chemical form of a trace metal as closely related
species, such as Pb2+ and PbCl+, yield only
a single stripping peak. Instead, trace metals
are divided into
several “operationally defined” cate- gories that have environmental significance.
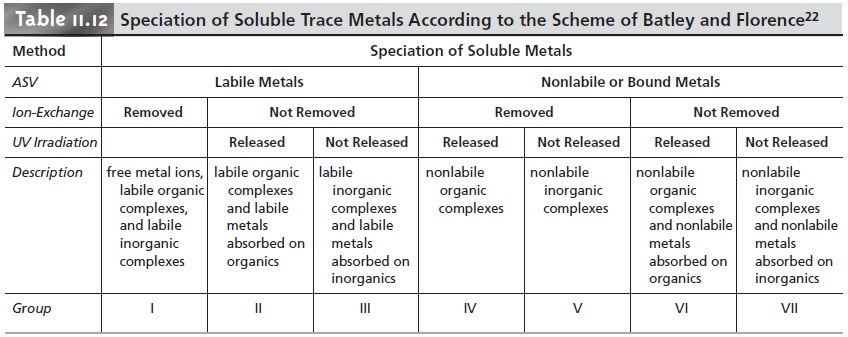
Several speciation schemes have been developed, but we
consider only the speciation scheme proposed
by Batley and Florence,
which uses a combination of anodic stripping voltammetry with ion-exchange and UV irradiation to divide solu- ble trace metals into seven classes.
Anodic stripping voltammetry in a pH 4.8 acetic acid buffer is used to distinguish labile metals present
as hydrated ions,
weakly bound complexes, or weakly adsorbed
on colloidal surfaces
from those metals
that are bound in stronger complexes
or strongly adsorbed.
Only those metals that are hydrated, weakly bound, or weakly adsorbed
deposit at the electrode. Ion exchange
and UV irradiation are used
to further subdivide the trace metals.
A Chelex-100 ion-exchange resin is used to distinguish between ionic metals and strongly
bound metals, whereas UV radiation is used to separate metals
bound to organic
and inor- ganic phases.
Table 11.12 shows
how trace metals
are divided into
seven classes using these three experimental techniques. The analysis
of sea water samples, for example, showed that cadmium, copper, and lead were primarily present as labile organic
complexes or as labile adsorbates on organic colloids (see group II, Table 11.12).
Differential pulse polarography and stripping voltammetry have been applied to the analysis of trace metals in airborne particulates, incinerator fly ash, rocks, minerals, and sediments.
The trace metals,
of course, must
be brought into
solution by digesting or extracting before
the voltammetric analysis.
Amperometric sensors also
are used to analyze environmental samples. For ex- ample, the dissolved O2 sensor described earlier
is routinely used for the determina-
tion of dissolved oxygen and
biochemical oxygen demand,
or BOD, in waters and wastewaters. The latter test,
which is a measure of the amount
of oxygen required by aquatic bacteria during
the decomposition of organic matter,
is of importance in
evaluating the efficiency of wastewater treatment plants and in monitoring organic pollution in natural
waters. A high BOD corresponds to a high concentration of or-
ganic material that may seriously
deplete the level of dissolved
oxygen in the water.
Other amperometric sensors
have been developed to monitor anionic
surfactants in water, and
CO2, H2SO4, and
NH3 in atmospheric gases.
Clinical Samples
Differential pulse
polarography and stripping voltammetry have been used to determine the concentration of trace metals
in a variety of matrices, including blood,
urine, and tissue
samples. The determination of lead in blood is of
considerable interest due to concerns
about lead poisoning. Because the concen- tration of lead in blood is so small,
anodic stripping voltammetry frequently is the method of choice. The
analysis is complicated, however, by the
presence of pro- teins that may adsorb at the surface of the mercury
electrode, inhibiting either the
deposition or stripping of lead.
In addition, proteins
may prevent the electrodepo-
sition of lead through the formation of stable, nonlabile complexes. For these
rea- sons samples of whole blood
must be digested
or ashed before
the analysis. Differ- ential pulse polarography is one of the few techniques that can be used for the
routine quantitative analysis
of drugs in biological fluids
at concentrations of less
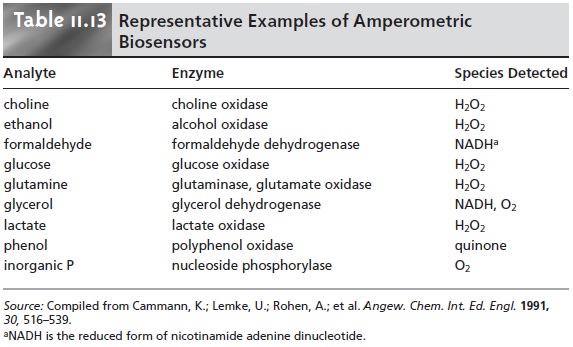
than 10–6 M.23 Amperometric sensors
based on enzyme
catalysts also have
wide ap- plicability.24 Table 11.13 provides a partial list of enzymatic amperometric sensors.
Miscellaneous Samples
Besides environmental and clinical samples, differential pulse polarography and stripping voltammetry have been used for the analysis of trace metals in other samples, including food, steels and other alloys, gasoline, gun- powder residues, and pharmaceuticals.
Voltammetry is also an important tool for the quantitative analysis of organics, particularly in the pharmaceutical industry, in which it is used to determine the concentration of drugs and vitamins in formula- tions.23 For example, voltammetric methods have been developed for the quantita- tive analysis of vitamin A, niacinamide, and riboflavin. When the compound of in- terest is not electroactive, it often can be derivatized to an electroactive form. One example is the differential pulse polarographic determination of sulfanilamide, in which it is converted into an electroactive azo dye by coupling with sulfamic acid and 1-naphthol.
Related Topics