Chapter: Modern Analytical Chemistry: Electrochemical Methods of Analysis
Controlled-Current Coulometry
Controlled-Current Coulometry
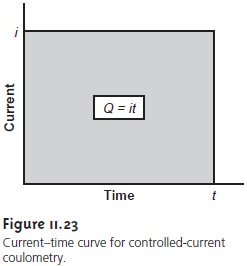
A second approach
to coulometry is to use a constant
current in place of a constant
potential (Figure 11.23).
Controlled-current coulometry, also
known as amperostatic coulometry or coulometric titrimetry, has two advantages over controlled-potential
coulometry. First, using
a constant current
makes for a more rapid
analysis since the current does not decrease
over time. Thus,
a typical analysis
time for controlled- current coulometry is less than 10 min, as opposed
to approximately 30–60 min for controlled-potential coulometry. Second, with a constant current
the total charge
is simply the product
of current and time (equation
11.24). A method for integrating the current–time curve, therefore, is not necessary.
Using a constant current does present two important experimental problems that must be solved
if accurate results
are to be obtained. First,
as electrolysis oc- curs the analyte’s concentration and, therefore, the current due to its oxidation
or reduction steadily decreases. To maintain a constant current
the cell potential must change until another
oxidation or reduction reaction can occur at the working
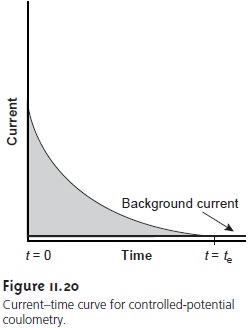
electrode. Unless the system is carefully designed, these secondary reac- tions will produce a current efficiency of less than 100%. The second problem
is the need for a method of determining when the analyte
has been exhaustively
electrolyzed. In controlled-potential coulometry this is signaled by a decrease in the current to a constant
background or residual
current (see Figure
11.20). In controlled-current coulometry, however, a constant
current continues to flow even when the analyte has been completely oxidized or reduced. A suitable means of determining the
end-point of the
reaction, te, is needed.
Maintaining Current Efficiency
To illustrate why
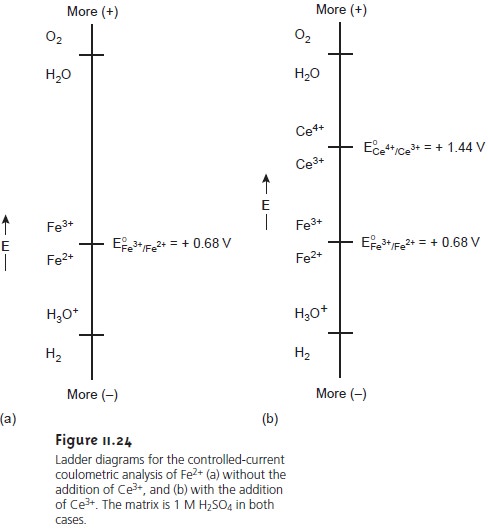
changing the working electrode’s potential can lead to less than 100% current efficiency, let’s consider the coulomet-
ric analysis for Fe2+ based on its oxidation to Fe3+ at a Pt working
electrode in 1 M
H2SO4.
Fe2+(aq)
< = = = = > Fe3+(aq)+ e–
The ladder diagram
for this system
is shown in Figure 11.24a.
Initially the potential of the working electrode remains nearly constant
at a level near the standard-state
potential for the
Fe3+/Fe2+ redox couple. As the
concentration of Fe2+ decreases,
however, the potential of the working electrode
shifts toward more positive values until another oxidation reaction can provide the
necessary current. Thus,
in this case the potential eventually increases to a level
at which the
oxidation of H2O occurs.
6H2O(l) < = = = = > O2(g)+ 4H3O+(aq)+ 4e–
Since the current
due to the oxidation of H3O+ does not contribute to the oxidation of Fe2+, the current
efficiency of the analysis is less than 100%. To maintain a 100%
current efficiency the products of any competing oxidation reactions must react
both rapidly and quantitatively with the remaining Fe2+. This may be accomplished, for example, by adding
an excess of Ce3+ to
the analytical solution
(Figure 11.24b). When the potential of the working
electrode shifts to a more positive potential, the first species to be oxidized
is Ce3+.
Ce3+(aq)
< = = = = > Ce4+(aq)+ e–
The Ce4+ produced at the working electrode rapidly mixes with the solution, where it reacts with any available Fe2+.
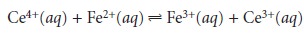 11.31
11.31
Combining these reactions gives the desired overall reaction of
Fe2+(aq)
< = = = = > Fe3+(aq)+ e–
In this manner,
a current efficiency of 100% is maintained. Furthermore, since the
concentration of Ce3+ remains at its initial
level, the potential of the working
elec- trode remains constant
as long as any Fe2+ is present.
This prevents other
oxidation reactions, such as that for H2O, from interfering with the analysis. A species, such as
Ce3+, which
is used to maintain 100% current efficiency, is called a mediator.
End Point Determination
Adding a mediator solves
the problem of maintaining
100% current efficiency, but does
not solve the
problem of determining when the analyte’s electrolysis is complete. Using
the same example, once all the
Fe2+ has been oxidized current continues to flow as a result
of the oxidation of Ce3+ and,
eventually, the oxidation of H2O. What is needed
is a means of indicating when the oxidation of Fe2+ is complete. In this respect it is convenient to treat a controlled-
current coulometric analysis as if electrolysis of the analyte
occurs only as a result
of its reaction with the mediator. A reaction between
an analyte and a mediator, such as that shown
in reaction 11.31,
is identical to that encountered in a redox
titration. Thus, the same end points
that are used in redox
titrimetry, such as visual indicators, and potentiometric and conductometric measurements, may be used to signal the
end point of a controlled-current coulometric analysis. For exam- ple, ferroin may be used to provide a visual end point for the Ce3+-mediated coulo- metric analysis for Fe2+.
Instrumentation
Controlled-current coulometry normally is carried
out using a galvanostat and an electrochemical cell consisting of a working
electrode and a counterelectrode. The working electrode, which often is constructed from Pt, is also
called the generator electrode since
it is where the mediator
reacts to generate
the species reacting with the analyte.
The counterelectrode is isolated from the analyti- cal solution by a salt bridge
or porous frit
to prevent its
electrolysis products from reacting with the analyte.
Alternatively, oxidizing or reducing the mediator can be
carried out externally, and the
appropriate products flushed
into the analytical solu- tion. Figure 11.25 shows one simple method by which oxidizing and reducing agents can be generated
externally. A solution
containing the mediator
flows under the influence of gravity into a small-volume electrochemical cell. The products gen- erated at the anode
and cathode pass through separate
tubes, and the appropriate
oxidizing or reducing reagent can be selectively delivered to the analytical solution.
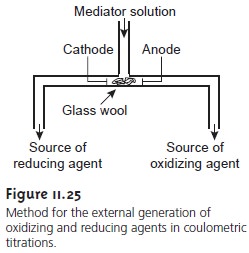
For example, external generation of Ce4+ can be obtained using
an aqueous solution of Ce3+ and the
products generated at the anode.
The other necessary instrumental component for controlled-current coulometry is an accurate clock for measuring
the electrolysis time, te, and a switch for starting and stopping the electrolysis. Analog clocks can read time to the nearest ±0.01
s, but the need
to frequently stop
and start the
electrolysis near the
end point leads
to a net uncertainty of ±0.1 s. Digital clocks provide a more accurate
measurement of time, with errors of ±1 ms being
possible. The switch
must control the flow of current and the
clock, so that an accurate
determination of the electrolysis time is possible.
Coulometric Titrations
Controlled-current coulometric methods commonly are called coulometric titrations because of their
similarity to conventional titrations. We already have noted, in discussing the
controlled-current coulometric determi-
nation of Fe2+, that the oxidation of Fe2+ by Ce4+ is identical to the reaction
used in a redox
titration. Other similarities between the two techniques also exist. Combining equations 11.23 and 11.24 and solving for the moles
of analyte gives
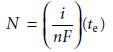 11.32
11.32
Compare this equation with the relationship between the moles of
strong acid, N, titrated with a
strong base of known concentration.
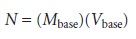
The titrant in a conventional titration is replaced
in a coulometric titration by a
constant-current source whose current is analogous to the titrant’s
molarity. The time needed
for an exhaustive electrolysis takes the
place of the
volume of titrant, and the switch for
starting and stopping the electrolysis serves
the same function as a buret’s stopcock.
Related Topics