Chapter: Medical Surgical Nursing: Assessment and Management of Patients With Hematologic Disorders
Sickle Cell Anemia
SICKLE
CELL ANEMIA
Sickle
cell anemia is a severe hemolytic anemia that results from inheritance of the
sickle hemoglobin gene. This gene causes the hemoglobin molecule to be
defective. The sickle hemoglobin (HbS) acquires a crystal-like formation when
exposed to low oxygen tension. The oxygen level in venous blood can be low
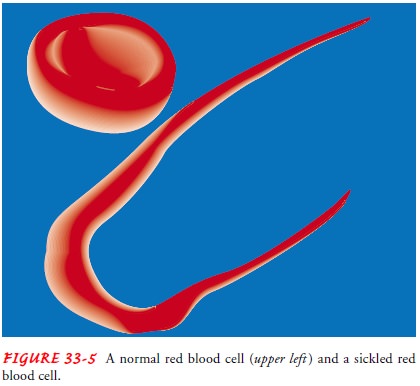
enough to cause this change; consequently, the RBC contain-ing (HbS) loses its
round, very pliable, biconcave disk shape and becomes deformed, rigid, and
sickle-shaped (Fig. 33-5). These long, rigid RBCs can adhere to the endothelium
of small ves-sels; when they pile up against each other, blood flow to a
re-gion or an organ may be reduced (Hoffman, et al., 2000). If ischemia or infarction
results, the patient may have pain, swelling, and fever. The sickling process
takes time; if the RBC is again exposed to adequate amounts of oxygen (eg, when
it travels through the pulmonary circulation) before the mem-brane becomes too
rigid, it can revert to a normal shape. For this reason, the “sickling crises”
are intermittent. Cold can aggravate
the
sickling process, because vasoconstriction slows the blood flow. Oxygen
delivery can also be impaired by an increased blood viscosity, with or without
occlusion due to adhesion of sickled cells; in this situation, the effects are
seen in larger ves-sels, such as arterioles.
The HbS gene is inherited in people of
African descent and to a lesser extent in people from the Middle East, the
Mediter-ranean area, and aboriginal tribes in India. Sickle cell anemia is the
most severe form of sickle cell disease. Less severe forms in-clude sickle cell
hemoglobin C (SC) disease, sickle cell hemo-globin D (SD) disease, and sickle
cell beta-thalassemia. The clinical manifestations and management are the same
as for sickle cell anemia. The term sickle
cell trait refers to the carrier state for SC diseases; it is the most
benign type of SC disease, in that less than 50% of the hemoglobin within an
RBC is HbS. However, in terms of genetic counseling, it is still an important
condition. If two people with sickle cell trait have children, the children may
inherit two abnormal genes. These children will produce only HbS and therefore
will have sickle cell anemia.
Clinical Manifestations
Symptoms
of sickle cell anemia vary and are only somewhat based on the amount of HbS.
Symptoms and complications result from chronic hemolysis or thrombosis. The
sickled RBCs have a short-ened life span. Patients are always anemic, usually
with hemoglo-bin values of 7 to 10 g/dL. Jaundice is characteristic and is
usually obvious in the sclerae. The bone marrow expands in childhood in a
compensatory effort to offset the anemia, sometimes leading to enlargement of
the bones of the face and skull. The chronic ane-mia is associated with
tachycardia, cardiac murmurs, and often an enlarged heart (cardiomegaly).
Dysrhythmias and heart failure may occur in adults.
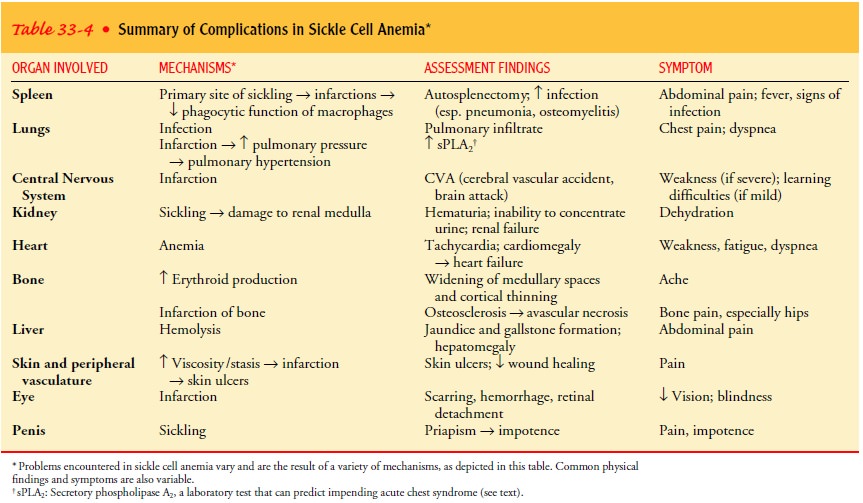
Virtually
any organ may be affected by thrombosis, but the pri-mary sites involve those
areas with slowed circulation, such as the spleen, lungs, and central nervous
system. All the tissues and or-gans are constantly vulnerable to
microcirculatory interruptions by the sickling process and therefore are susceptible
to hypoxic damage or true ischemic necrosis. Patients with sickle cell anemia
are unusually susceptible to infection, particularly pneumonia and
osteomyelitis. Complications of sickle cell anemia include infec-tion, stroke,
renal failure, impotence, heart failure, and pulmonary hypertension. Table 33-4
summarizes the complications resulting from sickle cell anemia.
SICKLE CELL CRISIS
There
are three types of sickle cell crisis in the adult population. The most common
is the very painful sickle crisis,
which results from tissue hypoxia and necrosis due to inadequate blood flow to
a specific region of tissue or organ. Aplastic
crisis results from in-fection with the human parvovirus. The hemoglobin
level falls rapidly and the marrow cannot compensate, as evidenced by an
absence of reticulocytes. Sequestration
crisis results when other or-gans pool the sickled cells. Although the
spleen is the most com-mon organ responsible for sequestration in children, by
10 years of age most children with sickle cell anemia have had a splenic
in-farction and the spleen is then no longer functional (autosplenec-tomy). In
adults, the common organs involved in sequestration are the liver and, more
seriously, the lungs.
ACUTE CHEST SYNDROME
Acute chest syndrome is manifested by a rapidly falling hemoglo-bin level, tachycardia, fever, and bilateral infiltrates seen on the chest x-ray. These signs often mimic infection; in fact, recent studies have identified infection as a major cause of acute chest syndrome (Vichinsky, et al., 2000). Another common cause is pulmonary fat embolism. Increased secretory phospholipase A2 concentration has been identified as a predictor of impending acute chest syndrome; the increased amounts of free fatty acids can cause increased permeability of the pulmonary endothelium and leakage of the pulmonary capillaries. Although this syndrome is potentially lethal, prompt intervention can result in a favorable outcome.
Assessment and Diagnostic Findings
The
patient with sickle cell trait usually has a normal hemoglobin level, a normal
hematocrit, and a normal blood smear. In contrast, the patient with sickle cell
anemia has a low hematocrit and sick-led cells on the smear. The diagnosis is
confirmed by hemoglobin electrophoresis.
Prognosis
Patients
with sickle cell anemia are usually diagnosed in child-hood, because they
become anemic in infancy and begin to have sickle cell crises at 1 or 2 years
of age. Some children die in the first years of life, typically from infection,
but the use of antibiotics and parent teaching have greatly improved the
outcomes for these chil-dren. However, with current management strategies, the
average life expectancy is still suboptimal, at 42 years. Young adults are
often forced to live with multiple, often severe, complications from their
disease. In some patients, the symptoms and compli-cations diminish by 30 years
of age; these patients live into the sixth decade or longer. At this time,
there is no way to predict which patients will fall into this subgroup.
Medical Management
Treatment
for sickle cell anemia is the focus of continued research (Steinberg, 1999).
Many trials of medications that have antisick-ling properties are being
conducted, as is research using antiadhe-sion treatment for vasoocclusive
crises. However, aside from the equally important aggressive management of
symptoms and com-plications, currently there are only three primary treatment
modalities for sickle cell diseases: BMT, hydroxyurea, and long-term RBC
transfusion.
BMT
offers the potential for cure for this disease. However, this treatment
modality is available to only a small subset of the patient population, because
of either the lack of a compatible donor or the severe organ (eg, renal, liver,
lung) damage already present in the patient.
PHARMACOLOGIC THERAPY
Hydroxyurea
(Hydrea), a chemotherapy agent, has been shown to be effective in increasing
hemoglobin F levels in patients with sickle cell anemia, thereby decreasing the
permanent formation of sickled cells. Patients who receive hydroxyurea appear
to have fewer painful episodes of sickle cell crisis, a lower incidence of
acute chest syndrome, and less need for transfusions (Ferster et al., 2001).
However, whether hydroxyurea can prevent or reverse ac-tual organ damage
remains unknown. Side effects of hydroxyurea include chronic suppression of WBC
formation, teratogenesis, and potential for later development of a malignancy.
Patient re-sponse to the medication varies significantly. The incidence and
severity of side effects are also highly variable within a dose range. Some
patients have toxicity when receiving a very small dose (5 mg/kg per day),
whereas others have little toxicity with a much higher dose (35 mg/kg per day).
More research is needed to iden-tify specific patient subgroups that are more
likely to respond to this medication.
TRANSFUSION THERAPY
Chronic
transfusions with RBCs have been shown to be highly ef-fective in several
situations: in an acute exacerbation of anemia (eg, aplastic crisis), in the
prevention of severe complications from anesthesia and surgery, and in
improving the response to infection (when it results in exacerbated anemia)
(Ohene-Frempong, 2001). Chronic transfusions have also been shown to be effective
in di-minishing episodes of sickle cell crisis in pregnant women; how-ever,
these transfusions have not been shown to improve fetal survival. Transfusion
therapy may be effective in preventing com-plications from sickle cell disease.
Although controversial, some data support the use of chronic transfusions in
patients with cere-bral ischemic injury (as seen on magnetic resonance imaging
[MRI] or Doppler studies) to prevent more severe injury (eg, CVA). More than
50% of asymptomatic patients have some cerebral ischemia documented by MRI. In
a recent study (Adams, 2000), chronic transfusion with RBCs resulted in a 90%
reduction of stroke in children at risk for this complication, as demonstrated
by elevated blood viscosity on transcranial Doppler ultrasonography.
Transfu-sions may also be useful in the management of severe cases of acute
chest syndrome.
The
risk of complications from transfusion is important to con-sider. These risks
include iron overload, which necessitates chronic chelation therapy (see MDS
Nursing Management); poor venous access, which necessitates a vascular access
device (and its attendant risk for infection or thrombosis); infections
(hepatitis, human immunodeficiency virus [HIV]); and alloimmunization from
re-peated transfusions. Another complication from transfusion is the increased
viscosity of blood before the concentration of hemoglo-bin S is reduced.
Exchange transfusion (in which the patient’s own blood is removed and replaced
via transfusion) may be performed to diminish the risk of increasing the
viscosity excessively; the ob-jective is to reduce the hematocrit to less than
30%, with transfu-sions supplying more than 80% of the patient’s blood volume.
Finally, it is important to consider the significant financial cost of an
aggressive transfusion and chelation program.
Patients
with sickle cell anemia require daily folic acid replace-ments to maintain the
supply required for increased erythropoiesis from hemolysis. Infections must be
treated promptly with appro-priate antibiotics; infection remains a major cause
of death in these patients.
Acute
chest syndrome is managed by prompt initiation of an-tibiotic therapy.
Incentive spirometry has been shown to decrease the incidence of pulmonary
complications significantly. In severe cases, bronchoscopy may be required to
identify the source of pulmonary disease. Fluid restriction may be more
beneficial than aggressive hydration. Corticosteroids may also be useful.
Trans-fusions reverse the hypoxia and decrease the level of secretory
phospholipase A2. Pulmonary function
should be monitored reg-ularly to detect pulmonary hypertension early, when
therapy (hydroxyurea, transfusions, or transplantation) may have a posi-tive
impact.
Because
repeated blood transfusions are necessary, patients may develop multiple
autoantibodies, making cross-matching difficult. In this patient population, a
hemolytic transfusion reaction (see later discussion) may mimic the signs and
symptoms of a sickle cell cri-sis. The classic distinguishing factor is that,
with a hemolytic trans-fusion reaction, the patient becomes more anemic after
being transfused. These patients need very close observation. Further
transfusion is avoided if possible until the hemolytic process abates. If
possible, the patient is supported with corticosteroids (Pred-nisone),
intravenous immunoglobulin (IVIG; Gammagard, Sando-globulin, Venoglobulin), and
erythropoietin (Epogen, Procrit).
SUPPORTIVE THERAPY
Supportive
care is equally important. A significant issue is pain management. The
incidence of painful sickle cell crises is highly variable; many patients have
pain on a daily basis. The severity of the pain may not be enough to cause the
patient to seek assistance from health care providers but severe enough to
interfere with the ability to work and function within the family. Acute pain
episodes tend to be self-limited, lasting hours to days. If the patient cannot
manage the pain at home, intervention is frequently sought in the acute care
setting, usually at an urgent care facility or emergency department. Adequate
hydration is important during a painful sickling episode. Oral hydration is
acceptable if the patient can maintain adequate amounts of fluids; intravenous
hydration with dextrose 5% in water (D5W) or dextrose 5% in 0.25 normal saline
solution (3 L/m2/24 hours) is usually
required for sickle crisis. Sup-plemental oxygen may also be needed.
The
use of medication to relieve pain is important. Aspirin is very useful in
diminishing mild to moderate pain; it also diminishes inflamma-tion and
potential thrombosis (due to its ability to diminish platelet adhesion).
Nonsteroidal anti-inflammatory drugs (NSAIDs) are useful for moderate pain or
in combination with opioid analgesics. Although no tolerance develops with
NSAIDs, a “ceiling effect” does develop whereby an increase in dosage does not
increase anal-gesia. NSAID use must be carefully monitored, because these
med-ications can precipitate renal dysfunction. When opioid analgesics are
used, morphine is the medication of choice for acute pain. Patient-controlled
analgesia is frequently used.
Chronic
pain increases in incidence as the patient ages. Here, the pain is caused by
complications from the sickling, such as avascular necrosis of the hip. With
chronic pain management, the principal goal is to maximize functioning; pain
may not be com-pletely eliminated without sacrificing function. This concept
may be difficult for patients to accept; they may need repeated expla-nations
and support from nonjudgmental health care providers. Nonpharmacologic
approaches to pain management are crucial in this setting. Examples include
physical and occupational therapy, physiotherapy (including the use of heat,
massage, and exercise), cognitive and behavioral intervention (including
distraction, relaxation, and motivational therapy), and support groups.
Working
with patients who have multiple episodes of severe pain can be challenging. It
is important for health care providers to realize that patients with sickle
cell disease must face a lifelong experience with severe and unpredictable
pain. Such pain is dis-ruptive to the person’s level of functioning, including
social func-tioning, and may result in a feeling of helplessness. Patients with
inadequate social support systems may have more difficulty cop-ing with chronic
pain.
Related Topics