Chapter: Modern Analytical Chemistry: Chromatographic and Electrophoretic Methods
Gas Chromatography: Sample Introduction
Sample Introduction
Three considerations determine how samples are introduced to the gas chromato-
graph. First, all
constituents injected into
the GC must
be volatile. Second,
the ana- lytes must be present
at an appropriate concentration. Finally,
injecting the sample must not degrade the separation.
Preparing a Volatile Sample
Gas chromatography can be used to separate
analytes in complex matrices.
Not every sample that can potentially be analyzed by GC,
however, can be injected directly
into the instrument. To move through
the col- umn, the sample’s constituents must be volatile. Solutes of low volatility may be re- tained by the column and continue
to elute during the analysis
of subsequent sam- ples. Nonvolatile solutes condense on the column, degrading the column’s
performance.
Volatile analytes can
be separated from
a nonvolatile matrix
using any of the
extraction techniques. Liquid–liquid extractions, in which analytes are extracted
from an aqueous matrix into methylene chloride
or other or- ganic
solvent, are commonly
used. Solid-phase extractions also are used to remove unwanted matrix constituents.
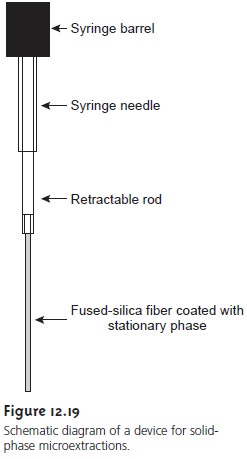
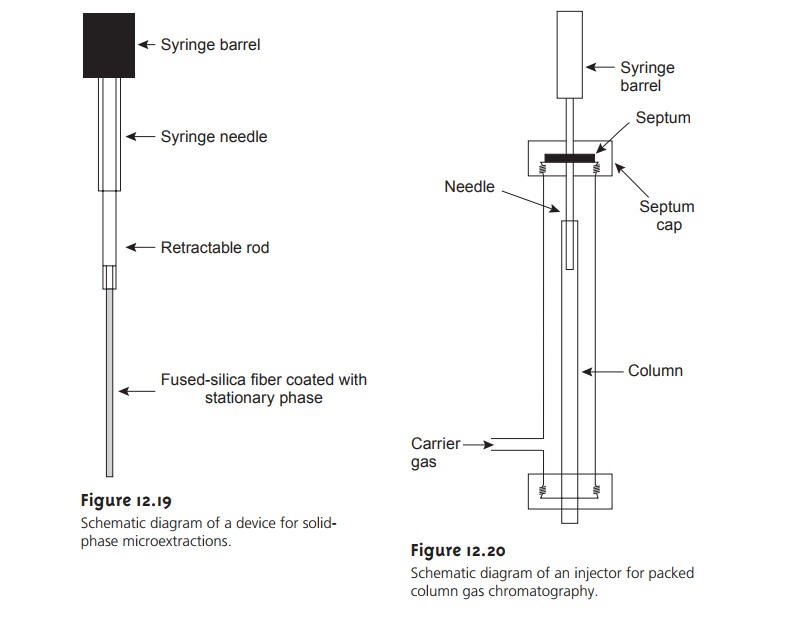
An attractive approach to isolating analytes is a solid-phase microextraction (SPME). In one approach, which is illustrated in Figure 12.19,
a fused silica
fiber is placed inside
a syringe needle.
The fiber, which
is coated with a thin organic film, such as polydimethyl siloxane,
is lowered into the sample by depressing a plunger and is exposed to the sample for a predetermined time. The fiber is then withdrawn
into the needle and transferred to the gas chromatograph for analysis.
Volatile analytes also can be separated from a liquid matrix using a purge and
trap or by headspace sampling. In a purge
and trap, an inert
gas, such as He or N2, is bubbled through
the sample, purging
the volatile compounds. These compounds are swept through
a trap packed with an absorbent
material, such as Tenax, where
they are collected. Heating the trap and back flush-
ing with carrier gas transfers
the volatile compounds
to the gas chromatograph. In headspace sampling the sample is placed in a closed
vial with an overlying air space. After
allowing time for the volatile
analytes to equilibrate between the sample and the overlying air,
a portion of the vapor
phase is sampled
by syringe and
in- jected into the gas chromatograph.
Thermal desorption is used to release volatile
analytes from solids.
A portion of the
solid is placed
in a glass-lined, stainless steel
tube and held in place
with plugs of glass
wool. After purging
with carrier gas to remove
O2 (which could
lead to oxida- tion reactions when heating
the sample), the sample is heated. Volatile
analytes are swept from the tube by the carrier gas and carried
to the GC. To maintain
efficiency the solutes often
are concentrated at the top
of the column
by cooling the
column inlet below room temperature, a process known
as cryogenic focusing.
Nonvolatile analytes must be chemically converted to a volatile derivative
before analysis. For
example, amino acids
are not sufficiently volatile to analyze directly by gas chromatography. Reacting an amino acid with 1-butanol and acetyl chloride produces an esterfied
amino acid. Subsequent treatment with
trifluoroacetic acid gives
the amino acid’s
volatile N-trifluoroacetyl-n-butyl ester
derivative.
Adjusting the Analyte’s Concentration
Analytes present at
concentrations too small to give an adequate signal
need to be concentrated before
analyzing. A side benefit of many of the extraction methods outlined earlier
is that they often concen- trate the analytes. Volatile organic
materials isolated from aqueous samples by a purge and trap, for
example, can be concentrated by as much
as 1000-fold.
When an analyte
is too concentrated, it is easy to overload the column, thereby seriously degrading the separation. In addition, the
analyte may be present at a con- centration level that exceeds
the detector’s linear
response. Dissolving the sample in a volatile solvent,
such as methylene chloride, makes its analysis feasible.
Injecting the Sample
To avoid
any precolumn loss
in resolution due
to band broadening, a sample of sufficient size must be introduced in a small
volume of mo- bile
phase. An example
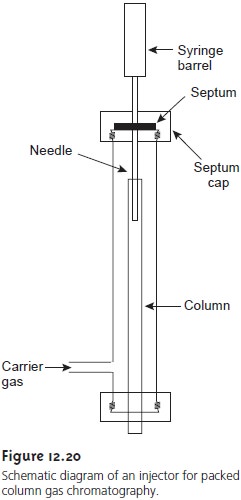
of a simple injection port for a packed column
is shown in Figure 12.20. Injections are made through
a rubber septum using a microliter sy- ringe. The injector block
is heated to a temperature that is at least 50 °C above
the sample component with the highest
boiling point. In this way rapid vaporization of the entire sample
is ensured.
Capillary columns require
the use of a special
injector to avoid overloading the column with sample. Several
capillary injectors are available, the most common
of which is a split/splitless injector.7 When used for a split injection only about
0.1–1% of the sample enters
the column, with the remainder carried off as waste. In a
splitless injection, which is useful for
trace analysis, the
column temperature is held
20–25 °C below
the solvent’s boiling
point. As the solvent enters
the column, it condenses, forming a barrier
that traps the solutes. After
allowing time for the solutes to concentrate, the column’s temperature is increased, and the separation begins. A splitless injection
allows a much higher percentage of the solutes
to enter the chromatographic column.
For samples that decompose easily,
an on-column injection may be necessary. In this method the
sample is injected on the column
without heating. The
column temperature is then increased, volatilizing the sample with as low a temperature as is practical.
Related Topics