Chapter: Basic & Clinical Pharmacology : Introduction
The Nature of Drugs
GENERAL PRINCIPLES OF PHARMACOLOGY
THE NATURE OF DRUGS
In
the most general sense, a drug may be defined as any substance that brings
about a change in biologic function through its chemical actions. In most
cases, the drug molecule interacts as an agonist
(activator) or antagonist (inhibitor)
with a specific mol-ecule in the biologic system that plays a regulatory role.
This target molecule is called a receptor.
In a very small number of cases, drugs known as chemical antagonists may interact directly with other drugs,
whereas a few drugs (osmotic agents) interact almost exclu-sively with water
molecules. Drugs may be synthesized within the body (eg, hormones) or may be chemicals not
synthesized in the body (ie, xenobiotics,
from the Greek xenos, meaning
“stranger”). Poisons are drugs that
have almost exclusively harmful effects.However, Paracelsus (1493–1541)
famously stated that “the dose makes the poison,” meaning that any substance
can be harmful if taken in the wrong dosage. Toxins are usually defined as poisons of biologic origin, ie,
synthesized by plants or animals, in contrast to inorganic poisons such as lead
and arsenic.To interact chemically with its receptor, a drug molecule must have
the appropriate size, electrical charge, shape, and atomic composition.
Furthermore, a drug is often administered at a loca-tion distant from its
intended site of action, eg, a pill given orally to relieve a headache. Therefore,
a useful drug must have the nec-essary properties to be transported from its
site of administration to its site of action. Finally, a practical drug should
be inactivated or excreted from the body at a reasonable rate so that its
actions will be of appropriate duration.
The Physical Nature of Drugs
Drugs
may be solid at room temperature (eg, aspirin, atropine), liquid (eg, nicotine,
ethanol), or gaseous (eg, nitrous oxide). These factors often determine the
best route of administration. The most common routes of administration are
described in Table 3–3. The various classes of organic compounds—carbohydrates,
proteins, lipids, and their constituents—are all represented in pharmacol-ogy.
As noted above, oligonucleotides, in the form of small seg-ments of RNA, have
entered clinical trials and are on the threshold of introduction into
therapeutics.
A
number of useful or dangerous drugs are inorganic elements, eg, lithium, iron,
and heavy metals. Many organic drugs are weak acids or bases. This fact has
important implications for the way they are handled by the body, because pH
differences in the vari-ous compartments of the body may alter the degree of
ionization of such drugs (see text that follows).
Drug Size
The
molecular size of drugs varies from very small (lithium ion, MW 7) to very
large (eg, alteplase [t-PA], a protein of MW 59,050). However, most drugs have
molecular weights between 100 and 1000. The lower limit of this narrow range is
probably set by the requirements for specificity of action. To have a good
“fit” to only one type of receptor, a drug molecule must be sufficiently unique
in shape, charge, and other properties, to prevent its bind-ing to other
receptors. To achieve such selective binding, it appears that a molecule should
in most cases be at least 100 MW units in size. The upper limit in molecular
weight is determined primarily by the requirement that drugs must be able to
move within the
body
(eg, from the site of administration to the site of action). Drugs much larger
than MW 1000 do not diffuse readily between compartments of the body (see
Permeation, in following text). Therefore, very large drugs (usually proteins)
must often be administered directly into the compartment where they have their
effect. In the case of alteplase, a clot-dissolving enzyme, the drug is
administered directly into the vascular compartment by intrave-nous or
intra-arterial infusion.
Drug Reactivity and Drug-Receptor Bonds
Drugs
interact with receptors by means of chemical forces or bonds. These are of
three major types: covalent,
electrostatic, and hydrophobic. Covalent
bonds are very strong and in many casesnot reversible under biologic
conditions. Thus, the covalent bond formed between the acetyl group of
acetylsalicylic acid (aspirin) and cyclooxygenase, its enzyme target in
platelets, is not readily broken. The platelet aggregation–blocking effect of
aspirin lasts long after free acetylsalicylic acid has disappeared from the
blood-stream (about 15 minutes) and is reversed only by the synthesis of new
enzyme in new platelets, a process that takes several days. Other examples of
highly reactive, covalent bond-forming drugs are the DNA-alkylating agents used
in cancer chemotherapy to disrupt cell division in the tumor.Electrostatic
bonding is much more common than covalent bonding in drug-receptor
interactions. Electrostatic bonds vary from relatively strong linkages between
permanently charged ionic molecules to weaker hydrogen bonds and very weak
induced dipole interactions such as van der Waals forces and similar
phe-nomena. Electrostatic bonds are weaker than covalent bonds.
Hydrophobic
bonds are usually quite weak and are probably important in the interactions of
highly lipid-soluble drugs with the lipids of cell membranes and perhaps in the
interaction of drugs with the internal walls of receptor “pockets.”
The
specific nature of a particular drug-receptor bond is of less practical
importance than the fact that drugs that bind through weak bonds to their
receptors are generally more selective than drugs that bind by means of very
strong bonds. This is because weak bonds require a very precise fit of the drug
to its receptor if an inter-action is to occur. Only a few receptor types are
likely to provide such a precise fit for a particular drug structure. Thus, if
we wished to design a highly selective short-acting drug for a particular
recep-tor, we would avoid highly reactive molecules that form covalent bonds
and instead choose a molecule that forms weaker bonds.
A
few substances that are almost completely inert in the chemical sense
nevertheless have significant pharmacologic effects. For example, xenon, an
“inert” gas, has anesthetic effects at ele-vated pressures.
Drug Shape
The
shape of a drug molecule must be such as to permit binding to its receptor site
via the bonds just described. Optimally, the drug’s shape is complementary to
that of the receptor site in the same way that a key is complementary to a
lock. Furthermore, the phenome-non of chirality
(stereoisomerism) is so common in biology that more than half of all useful
drugs are chiral molecules; that is, they can exist as enantiomeric pairs.
Drugs with two asymmetric centers have four diastereomers, eg, ephedrine, a
sympathomimetic drug. In most cases, one of these enantiomers is much more
potent than its mirror image enantiomer, reflecting a better fit to the
receptor mol-ecule. If one imagines the receptor site to be like a glove into
which the drug molecule must fit to bring about its effect, it is clear why a
“left-oriented” drug is more effective in binding to a left-hand receptor than
its “right-oriented” enantiomer.
The
more active enantiomer at one type of receptor site may not be more active at
another receptor type, eg, a type that may be responsible for some other
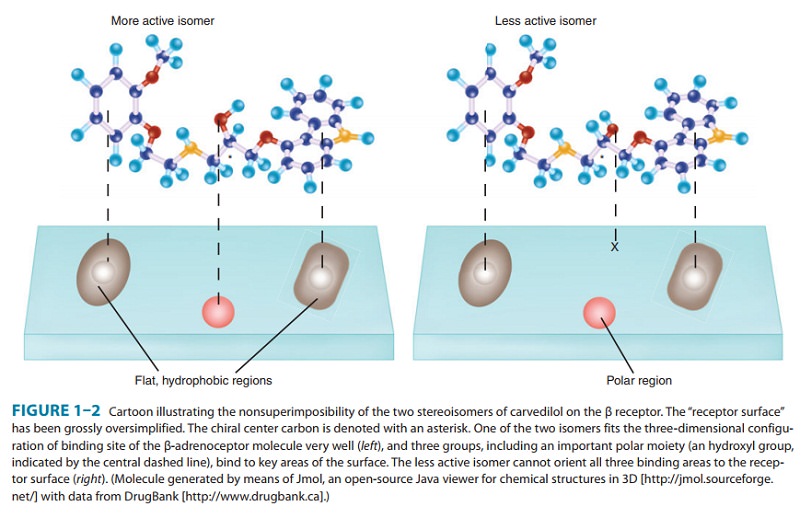
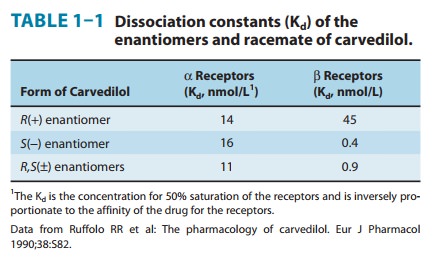
effect. For example, carvedilol, a drug that interacts with adrenoceptors, has
a single chiral center and thus two enantiomers (Figure 1–2, Table 1–1). One of
these enantiomers, the (S)(−) isomer, is a potent β-receptor blocker. The
(R)(+) isomer is 100-fold weaker at the β receptor. However,
theisomers are approximately equipotent as α-receptor blockers. Ketamine is an intravenous
anesthetic. The (+)
enantiomer is a more potent anesthetic and is less toxic than the (−) enantiomer.
Unfortunately, the drug is still used as the racemic mixture.
Finally,
because enzymes are usually stereoselective, one drug enantiomer is often more
susceptible than the other to drug-metabolizing enzymes. As a result, the
duration of action of one enantiomer may be quite different from that of the
other. Similarly, drug transporters may be stereoselective.
Unfortunately,
most studies of clinical efficacy and drug elimi-nation in humans have been
carried out with racemic mixtures ofdrugs rather than with the separate
enantiomers. At present, only a small percentage of the chiral drugs used
clinically are marketed as the active isomer—the rest are available only as
racemic mixtures. As a result, many patients are receiving drug doses of which
50% is less active, inactive, or actively toxic. Some drugs are currently
available in both the racemic and the pure, active isomer forms. Unfortunately,
the hope that administration of the pure, active enantiomer would decrease
adverse effects relative to those pro-duced by racemic formulations has not
been firmly established. However, there is increasing interest at both the
scientific and the regulatory levels in making more chiral drugs available as
their active enantiomers
Rational Drug Design
Rational
design of drugs implies the ability to predict the appropri-ate molecular
structure of a drug on the basis of information about its biologic receptor.
Until recently, no receptor was known in suf-ficient detail to permit such drug
design. Instead, drugs were devel-oped through random testing of chemicals or
modification of drugs already known to have some effect . However, the
characterization of many receptors during the past three decades has changed
this picture. A few drugs now in use were developed through molecular design
based on knowledge of the three-dimen-sional structure of the receptor site.
Computer programs are now available that can iteratively optimize drug
structures to fit known receptors. As more becomes known about receptor
structure, ratio-nal drug design will become more common.
Receptor Nomenclature
The spectacular success of newer, more efficient ways to identify and characterize receptors has resulted in a variety of differing, and sometimes confusing, systems for naming them. This in turn has led to a number of suggestions regarding more rational methods of naming receptors. The interested reader is referred for details to the efforts of the International Union of Pharmacology (IUPHAR) Committee on Receptor Nomenclature andDrug Classification (reported in various issues of Pharmacological Reviews) and to Alexander SPH, Mathie A, Peters JA: Guide toreceptors and channels (GRAC), 4th edition. Br J Pharmacol 2009;158(Suppl 1):S1–S254.
Related Topics