Chapter: Basic & Clinical Pharmacology : Introduction to Autonomic Pharmacology
Functional Organization of Autonomic Activity
FUNCTIONAL ORGANIZATION OF
AUTONOMIC ACTIVITY
Autonomic function is integrated and regulated at many levels, from the CNS to the effector cells. Most regulation uses negative feedback, but several other mechanisms have been identified. Negative feedback is particularly important in the responses of the ANS to the administration of autonomic drugs.
Central Integration
At
the highest level—midbrain and medulla—the two divisions of the ANS and the
endocrine system are integrated with each other, with sensory input, and with
information from higher CNS centers, including the cerebral cortex. These
interactions are such that early investigators called the parasympathetic
system a trophotropic one (ie,
leading to growth) used to “rest and digest” and the sympathetic system an ergotropic one (ie, leading to energy
expenditure), which is activated for “fight or flight.” Although such terms
offer little insight into the mechanisms involved, they do provide simple
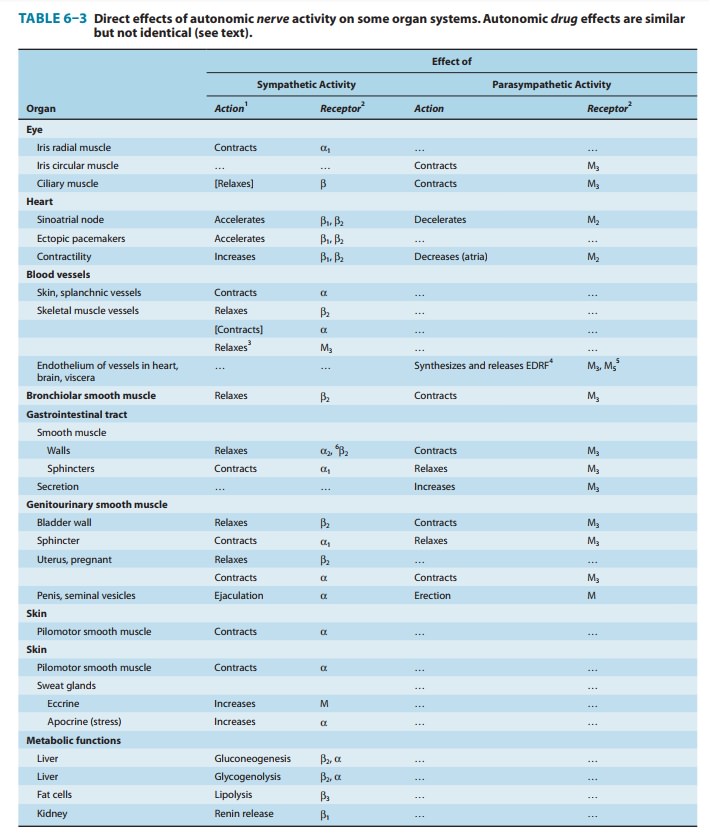
descrip-tions applicable to many of the actions of the systems (Table 6–3). For
example, slowing of the heart and stimulation of digestive activity are typical
energy-conserving and storing actions of the parasympathetic system. In
contrast, cardiac stimulation, increased blood sugar, and cutaneous
vasoconstriction are responses produced by sympathetic discharge that are
suited to fighting or surviving attack.
At
a more subtle level of interactions in the brain stem, medulla, and spinal
cord, there are important cooperative interactions between the parasympathetic
and sympathetic systems. For some organs, sensoryfibers associated with the
parasympathetic system exert reflex control over motor outflow in the
sympathetic system. Thus, the sensory carotid sinus baroreceptor fibers in the
glossopharyngeal nerve have a major influence on sympathetic outflow from the
vasomotor center. This example is described in greater detail in the following
text. Similarly, parasympathetic sensory fibers in the wall of the urinary
blad-der significantly influence sympathetic inhibitory outflow to that organ.
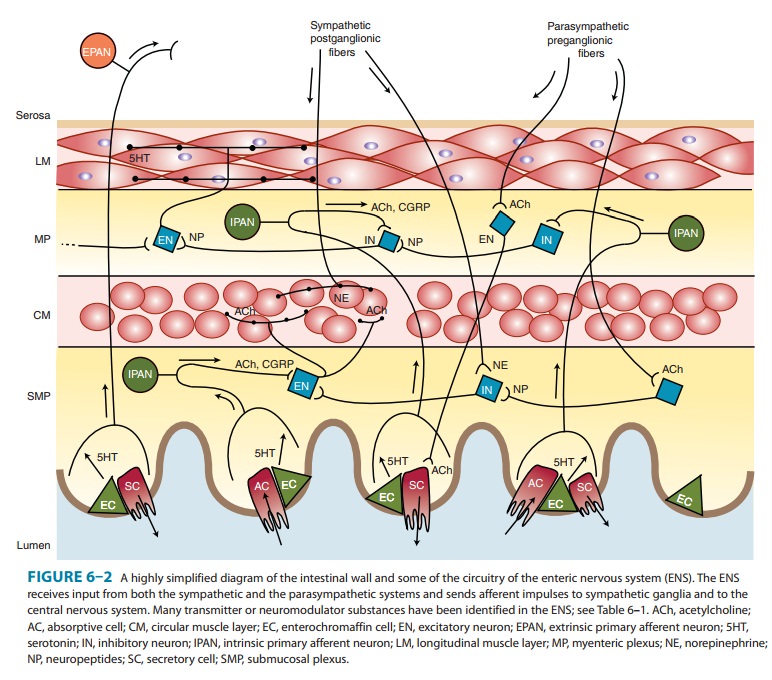
Within the ENS, sensory fibers from the wall of the gut synapse on both
preganglionic and postganglionic motor cells that control intesti-nal smooth
muscle and secretory cells (Figure 6–2).
Integration of Cardiovascular Function
Autonomic
reflexes are particularly important in understanding cardiovascular responses
to autonomic drugs. As indicated in Figure 6–7, the primary controlled variable
in cardiovascular func-tion is mean
arterial pressure. Changes in any variable contribut-ing to mean arterial
pressure (eg, a drug-induced increase in peripheral vascular resistance) evoke
powerful homeostatic second-ary
responses that tend to compensate for the directly evoked
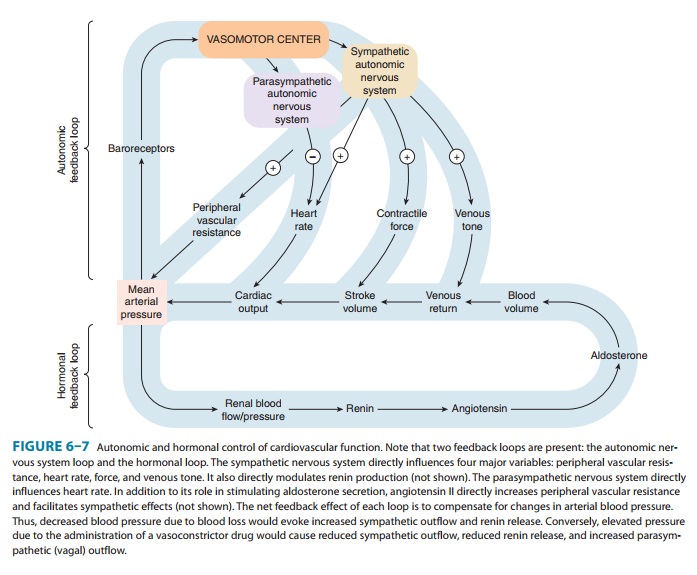
change.
The homeostatic response may be sufficient to reduce the change in mean
arterial pressure and to reverse the drug’s effects on heart rate. A slow
infusion of norepinephrine provides a useful example. This agent produces
direct effects on both vascular and cardiac muscle. It is a powerful
vasoconstrictor and, by increasing peripheral vascular resistance, increases
mean arterial pressure. In the absence of reflex control—in a patient who has
had a heart trans-plant, for example—the drug’s effect on the heart is also
stimulatory; that is, it increases heart rate and contractile force. However,
in a subject with intact reflexes, the negative feedback response to increased
mean arterial pressure causes decreased sympathetic out-flow to the heart and a
powerful increase in parasympathetic (vagus nerve) discharge at the cardiac
pacemaker. This response is mediated by increased firing by the baroreceptor
nerves of the carotid sinus and the aortic arch. Increased baroreceptor
activity causes the changes mentioned in central sympathetic and vagal outflow.
As a result, the net effect of
ordinary pressor doses of norepinephrine in a normalsubject is to produce a
marked increase in peripheral vascular resis-tance, an increase in mean
arterial pressure, and a consistent slowingof
heart rate. Bradycardia, the reflex compensatory response elicited by this
agent, is the exact opposite of the
drug’s direct action; yet it is completely predictable if the integration of
cardiovascular function by the ANS is understood.
Presynaptic Regulation
The
principle of negative feedback control is also found at the presynaptic level
of autonomic function. Important presynaptic feedback inhibitory control
mechanisms have been shown to exist at most nerve endings. A well-documented
mechanism involves the α2 receptor located on noradrenergic nerve terminals.
This receptor is activated by norepinephrine and similar molecules; activation
diminishes further release of norepinephrine from these nerve endings (Table
6–4). The mechanism of this G protein-mediated effect involves inhibition of
the inward calcium current that causes vesicular fusion and transmitter
release. Conversely, a presynaptic β receptor appears to facilitate the release of
norepi-nephrine from some adrenergic neurons. Presynaptic receptorsthat respond
to the primary transmitter substance released by the nerve ending are called autoreceptors. Autoreceptors are
usually inhibitory, but in addition to the excitatory β receptors on nor-adrenergic fibers, many
cholinergic fibers, especially somatic motor fibers, have excitatory nicotinic
autoreceptors.

Control
of transmitter release is not limited to modulation by the transmitter itself.
Nerve terminals also carry regulatory recep-tors that respond to many other
substances. Such heteroreceptors may
be activated by substances released from other nerve termi-nals that synapse
with the nerve ending. For example, some vagal fibers in the myocardium synapse
on sympathetic noradrenergic nerve terminals and inhibit norepinephrine
release. Alternatively, the ligands for these receptors may diffuse to the receptors
from the blood or from nearby tissues. Some of the transmitters and receptors
identified to date are listed in Table 6–4. Presynaptic regulation by a variety
of endogenous chemicals probably occurs in all nerve fibers.
Postsynaptic Regulation
Postsynaptic
regulation can be considered from two perspectives: modulation by previous
activity at the primary receptor (which may up- or down-regulate receptor
number or desensitize receptors;), and modulation by other simultaneous events.
The
first mechanism has been well documented in several receptor-effector systems.
Up-regulation and down-regulation are known to occur in response to decreased
or increased activation, respectively, of the receptors. An extreme form of
up-regulation occurs after denervation of some tissues, resulting in denervationsupersensitivity of the
tissue to activators of that receptor type. Inskeletal muscle, for example,
nicotinic receptors are normally restricted to the end-plate regions underlying
somatic motor nerve terminals. Surgical denervation results in marked
proliferation of nicotinic cholinoceptors over all parts of the fiber,
including areas not previously associated with any motor nerve junctions. A
phar-macologic supersensitivity related to denervation supersensitivity occurs
in autonomic effector tissues after administration of drugs that deplete
transmitter stores and prevent activation of the post-synaptic receptors for a
sufficient period of time. For example, prolonged administration of large doses
of reserpine, a norepi-nephrine depleter, can cause increased sensitivity of
the smooth muscle and cardiac muscle effector cells served by the depleted
sympathetic fibers.
The
second mechanism involves modulation of the primary transmitter-receptor event
by events evoked by the same or other transmitters acting on different
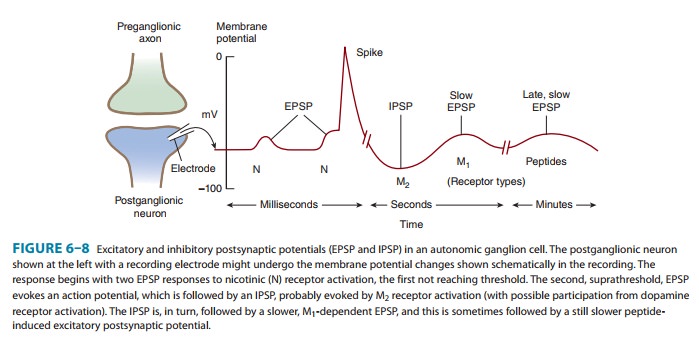
postsynaptic receptors. Ganglionic transmission is a good example of this
phenomenon (Figure 6–8). The postganglionic cells are activated (depolarized)
as a result of binding of an appropriate ligand to a neuronal nicotinic (NN)
ace-tylcholine receptor. The resulting fast excitatory postsynapticpotential (EPSP) evokes a propagated action
potential if thresholdis reached. This event is often followed by a small and
slowly devel-oping but longer-lasting hyperpolarizing afterpotential—a slow inhibitory postsynaptic potential (IPSP). This
hyperpolarizationinvolves opening of potassium channels by M2
cholinoceptors. The IPSP is followed by a small, slow excitatory postsynaptic
potential caused by closure of potassium channels linked to M1
cholinocep-tors. Finally, a late, very slow EPSP may be evoked by peptides
released from other fibers. These slow potentials serve to modulate the
responsiveness of the postsynaptic cell to subsequent primary excitatory
presynaptic nerve activity.
Related Topics