Chapter: Basic & Clinical Pharmacology : Introduction to the Pharmacology of Central Nervous System (CNS) Drugs
Central Neurotransmitters
CENTRAL NEUROTRANSMITTERS
A
vast number of small molecules have been isolated from the brain, and studies
using a variety of approaches suggest that the agents listed in Table 21–2 are
neurotransmitters. A brief sum-mary of the evidence for some of these compounds
follows.
Amino Acids
The amino acids of primary interest to the pharmacologist fall into two categories: the acidic amino acid glutamate and the neu-tral amino acids glycine and GABA. All these compounds are present in high concentrations in the CNS and are extremely potent modifiers of neuronal excitability.
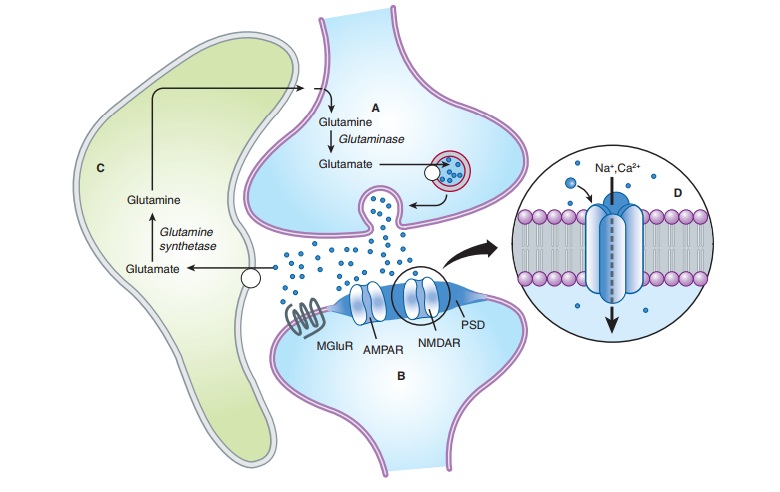
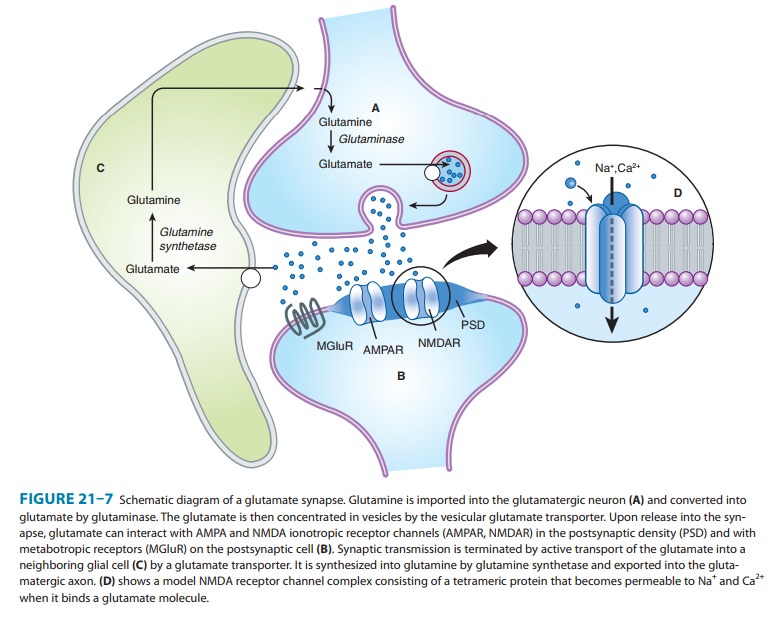
A. Glutamate
Excitatory
synaptic transmission is mediated by glutamate, which is present in very high
concentrations in excitatory synaptic vesi-cles (∼100 mM). Glutamate is released into the
synaptic cleft by Ca2+-dependent exocytosis (Figure 21–7). The released glutamate acts
on postsynaptic glutamate receptors and is cleared by gluta-mate transporters
present on surrounding glia. In glia, glutamate is converted to glutamine by
glutamine synthetase, released fromthe glia, taken up by the nerve terminal,
and converted back to glutamate by the enzyme glutaminase. The high
concentration of glutamate in synaptic vesicles is achieved by the vesicular gluta-mate transporter (VGLUT).
Almost
all neurons that have been tested are strongly excited by glutamate. This excitation
is caused by the activation of both ionotropic and metabotropic receptors,
which have been exten-sively characterized by molecular cloning. The ionotropic
recep-tors can be further divided into three subtypes based on the action of
selective agonists: α-amino-3-hydroxy-5-methylisoxazole-4-propionicacid
(AMPA), kainicacid(KA), andN-methyl-D-aspartate
All the ionotropic receptors are composed of
four sub-units. AMPA receptors, which are present on all neurons, are
heterotetramers assembled from four subunits (GluA1–GluA4). The majority of
AMPA receptors contain the GluA2 subunit and are permeable to Na+ and K+, but not to Ca2+. Some AMPA
recep-tors, typically present on inhibitory interneurons, lack the GluA2
subunit and are also permeable to Ca2+.
Kainate
receptors are not as uniformly distributed as AMPA receptors, being expressed
at high levels in the hippocampus, cere-bellum, and spinal cord. They are
formed from a number of sub-unit combinations (GluK1–GluK5). Although GluK4 and
GluK5 are unable to form channels on their own, their presence in the receptor
changes the receptor’s affinity and kinetics. Similar to AMPA receptors,
kainate receptors are permeable to Na+ and K+ and in some subunit
combinations can also be permeable to Ca2+.
NMDA
receptors are as ubiquitous as AMPA receptors, being present on essentially all
neurons in the CNS. All NMDA receptors require the presence of the subunit
GluN1. The channel also contains one or two NR2 subunits (GluN2A–GluN2D).
Unlike
AMPA and kainate receptors, all NMDA receptors are highly permeable to Ca2+ as well as to Na+ and K+. NMDA recep-tor
function is controlled in a number of intriguing ways. In addi-tion to
glutamate binding, the channel also requires the binding of glycine to a
separate site. The physiologic role of glycine bind-ing is unclear because the
glycine site appears to be saturated at normal ambient levels of glycine.
Another key difference between AMPA and kainate receptors on the one hand, and
NMDA recep-tors on the other, is that AMPA and kainate receptor activation
results in channel opening at resting membrane potential, whereas NMDA receptor
activation does not. This is due to the voltage-dependent block of the NMDA
pore by extracellular Mg2+. When the neuron is
strongly depolarized, as occurs with intense activa-tion of the synapse or by
activation of neighboring synapses, Mg2+ is expelled and the
channel opens. Thus, there are two require-ments for NMDA receptor channel
opening: Glutamate must bind the receptor and the membrane must be depolarized.
The rise in intracellular Ca2+ that accompanies
channel opening results in a long-lasting enhancement in synaptic strength that
is referred to as long-term potentiation
(LTP). The change can last for many hours or even days and is generally
accepted as an important cellular mechanism underlying learning and memory.
The
metabotropic glutamate receptors are G protein-coupled receptors that act
indirectly on ion channels via G proteins. Metabotropic receptors
(mGluR1–mGluR8) have been divided into three groups (I, II, and III). A variety
of agonists and antago-nists have been developed that interact selectively with
the differ-ent groups. Group I receptors are typically located postsynaptically
and are thought to cause neuronal excitation by activating a non-selective
cation channel. These receptors also activate phospholi-pase C, leading to
inositol trisphosphate-mediated intracellular Ca2+ release. In contrast,
group II and group III receptors are typically located on presynaptic nerve
terminals and act as inhib-itory autoreceptors. Activation of these receptors
causes the inhi-bition of Ca2+ channels, resulting
in inhibition of transmitter release. These receptors are activated only when
the concentration of glutamate rises to high levels during repetitive
stimulation of the synapse. Activation of these receptors causes the inhibition
of adenylyl cyclase and decreases cAMP generation.
The
postsynaptic membrane at excitatory synapses is thickened and referred to as
the postsynaptic density (PSD;
Figure 21–7). This is a highly complex structure containing glutamate
receptors, signaling proteins, scaffolding proteins, and cytoskeletal proteins.
A typical excitatory synapse contains AMPA receptors, which tend to be located
toward the periphery, and NMDA receptors, which are concentrated in the center.
Kainate receptors are present at a subset of excitatory synapses, but their
exact location is unknown. Metabotropic glutamate receptors (group I), which
are localized just outside the postsynaptic density, are also present at some
excitatory synapses.
B. GABA and Glycine
Both
GABA and glycine are inhibitory neurotransmitters, which are typically released
from local interneurons. Interneurons that release glycine are restricted to
the spinal cord and brainstem, whereas interneurons releasing GABA are present
throughout the CNS, including the spinal cord. It is interesting that some
interneurons in the spinal cord can release both GABA and gly-cine. Glycine
receptors are pentameric structures that are selec-tively permeable to Cl–.
Strychnine, which is a potent spinal cord convulsant and has been used in some
rat poisons, selectively blocks glycine receptors.
GABA
receptors are divided into two main types: GABAA and GABAB.
Inhibitory postsynaptic potentials in many areas of the brain have a fast and
slow component. The fast component is mediated by GABAA receptors
and the slow component by GABAB receptors. The difference in
kinetics stems from the dif-ferences in coupling of the receptors to ion
channels. GABAA receptors are ionotropic receptors and, like glycine
receptors, are pentameric structures that are selectively permeable to Cl–.
These receptors are selectively inhibited by picrotoxin and bicu-culline, both
of which cause generalized convulsions. A great many subunits for GABAA
receptors have been cloned; this accounts for the large diversity in the
pharmacology of GABAAreceptors, making them key targets for
clinically useful agents . GABAB receptors are metabotropic
receptors that are selectively activated by the antispastic drug baclofen.
These receptors are coupled to G proteins that, depending on their cellular
location, either inhibit Ca2+ channels or activate
K+ channels. The GABAB
component of the inhibitory postsyn-aptic potential is due to a selective
increase in K+ conductance. This inhibitory postsynaptic potential is
long-lasting and slow because the coupling of receptor activation to K+ channel open-ing is
indirect and delayed. GABAB receptors are localized to the
perisynaptic region and thus require the spillover of GABA from the synaptic
cleft. GABAB receptors are also present on the axon terminals of
many excitatory and inhibitory synapses. In this case, GABA spills over onto
these presynaptic GABAB receptors, inhibiting transmitter release by
inhibiting Ca2+ channels. In addition to their coupling to ion channels, GABAB
receptors also inhibit adenylyl cyclase and decrease cAMP generation.
Acetylcholine
Acetylcholine
was the first compound to be identified pharma-cologically as a transmitter in
the CNS. Eccles showed in the early 1950s that excitation of Renshaw cells by
motor axon collaterals in the spinal cord was blocked by nicotinic
antago-nists. Furthermore, Renshaw cells were extremely sensitive to nicotinic
agonists. These experiments were remarkable for two reasons. First, this early
success at identifying a transmitter for a central synapse was followed by
disappointment because it remained the sole central synapse for which the
transmitter was known until the late 1960s, when comparable data became
available for GABA and glycine. Second, the motor axon col-lateral synapse
remains one of the best-documented examples of a cholinergic nicotinic synapse
in the mammalian CNS, despite the rather widespread distribution of nicotinic
recep-tors as defined by in situ hybridization studies. Most CNS responses to
acetylcholine are mediated by a large family of G protein-coupled muscarinic
receptors. At a few sites, acetylcho-line causes slow inhibition of the neuron
by activating the M2 subtype of receptor, which opens potassium
channels. A far more widespread muscarinic action in response to acetylcho-line
is a slow excitation that in some cases is mediated by M1 receptors.
These muscarinic effects are much slower than either nicotinic effects on
Renshaw cells or the effect of amino acids. Furthermore, this M1
muscarinic excitation is unusual in that acetylcholine produces it by decreasing the membrane permea-bility to
potassium, ie, the opposite of conventional transmitter action.
A
number of pathways contain acetylcholine, including neu-rons in the
neostriatum, the medial septal nucleus, and the reticu-lar formation.
Cholinergic pathways appear to play an important role in cognitive functions,
especially memory. Presenile dementia of the Alzheimer type is reportedly
associated with a profound loss of cholinergic neurons. However, the
specificity of this loss has been questioned because the levels of other
putative transmitters, eg, somatostatin, are also decreased.
Monoamines
Monoamines
include the catecholamines (dopamine and norepi-nephrine) and
5-hydroxytryptamine. Although these compounds are present in very small amounts
in the CNS, they can be local-ized using extremely sensitive histochemical
methods. These path-ways are the site of action of many drugs; for example, the
CNS stimulants cocaine and amphetamine appear to act primarily at catecholamine
synapses. Cocaine blocks the reuptake of dopamine and norepinephrine, whereas
amphetamines cause presynaptic terminals to release these transmitters.
A. Dopamine
The
major pathways containing dopamine are the projection linking the substantia
nigra to the neostriatum and the projec-tion linking the ventral tegmental
region to limbic structures, particularly the limbic cortex. The therapeutic
action of the antiparkinsonism drug levodopa is associated with the former area
, whereas the therapeutic action of the antipsychotic drugs is thought to be
associated with the latter . Dopamine-containing neurons in the tubero-basal
ventral hypothalamus play an important role in regulating
hypothalamohypophysial function. Five dopamine receptors have been identified,
and they fall into two categories: D1-like (D1 and D5)
and D2-like (D2, D3, D4). All
dopamine receptors are metabotropic. Dopamine generally exerts a slow
inhibitory action on CNS neurons. This action has been best characterized on
dopamine-containing substantia nigra neurons, where D2-receptor
activation opens potassium channels via the Gi coupling protein.
B. Norepinephrine
Most
noradrenergic neurons are located in the locus caeruleus or the lateral
tegmental area of the reticular formation. Although the density of fibers
innervating various sites differs considerably, most regions of the CNS receive
diffuse nor-adrenergic input. All noradrenergic receptor subtypes are
metabotropic. When applied to neurons, norepinephrine can hyperpolarize them by
increasing potassium conductance. This effect is mediated by α2 receptors and has
been characterized most thoroughly on locus caeruleus neurons. In many regions
of the CNS, norepinephrine actually enhances excitatory inputs by both indirect
and direct mechanisms. The indirect mechanism involves disinhibition; that is,
inhibitory local cir-cuit neurons are inhibited. The direct mechanism involves
blockade of potassium conductances that slow neuronal dis-charge. Depending on
the type of neuron, this effect is medi-ated by either α1 or β receptors. Facilitation of excitatory
synaptic transmission is in accordance with many of the behav-ioral processes
thought to involve noradrenergic pathways, eg, attention and arousal.
C. 5-Hydroxytryptamine
Most
5-hydroxytryptamine (5-HT, serotonin) pathways originate from neurons in the
raphe or midline regions of the pons and upper brainstem. 5-HT is contained in
unmyelinated fibers thatdiffusely innervate most regions of the CNS, but the
density of the innervation varies. 5-HT acts on more than a dozen receptor subtypes.
Except for the 5-HT 3 receptor, all of these receptors are
metabotropic. The ionotropic 5-HT3 receptor exerts a rapid
excitatory action at a very limited number of sites in the CNS. In most areas
of the CNS, 5-HT has a strong inhibitory action. This action is mediated by
5-HT1A receptors and is associated with membrane hyperpolarization
caused by an increase in potassium conductance. It has been found that 5-HT1A
receptors and GABAB receptors activate the same population of
potassium channels. Some cell types are slowly excited by 5-HT owing to its
blockade of potassium channels via 5-HT 2 or 5-HT4
receptors. Both excitatory and inhibitory actions can occur on the same neuron.
It has often been speculated that 5-HT pathways may be involved in the hallucinations
induced by LSD (lysergic acid), since this compound can antagonize the
peripheral actions of 5-HT. However, LSD does not appear to be a 5-HT
antagonist in the CNS, and typical LSD-induced behavior is still seen in
animals after raphe nuclei are destroyed. Other proposed regula-tory functions
of 5-HT-containing neurons include sleep, tem-perature, appetite, and
neuroendocrine control.
Peptides
A
great many CNS peptides have been discovered that produce dramatic effects both
on animal behavior and on the activity of individual neurons. Many of the
peptides have been mapped with immunohistochemical techniques and include
opioid peptides (eg, enkephalins, endorphins), neurotensin, substance P,
soma-tostatin, cholecystokinin, vasoactive intestinal polypeptide,
neu-ropeptide Y, and thyrotropin-releasing hormone. As in the peripheral
autonomic nervous system, peptides often coexist with a conventional nonpeptide
transmitter in the same neuron. A good example of the approaches used to define
the role of these peptides in the CNS comes from studies on substance P and its
association with sensory fibers. Substance P is contained in and released from
small unmyelinated primary sensory neurons in the spinal cord and brainstem and
causes a slow excitatory postsyn-aptic potential in target neurons. These
sensory fibers are known to transmit noxious stimuli, and it is therefore
surprising that— although substance P receptor antagonists can modify responses
to certain types of pain—they do not block the response. Glutamate, which is
released with substance P from these syn-apses, presumably plays an important
role in transmitting pain stimuli. Substance P is certainly involved in many
other functions because it is found in many areas of the CNS that are unrelated
to pain pathways.
Many
of these peptides are also found in peripheral structures, including peripheral
synapses.
Nitric Oxide
The
CNS contains a substantial amount of nitric oxide syn-thase (NOS) within
certain classes of neurons. This neuronal NOS is an enzyme activated by
calcium-calmodulin, and acti-vation of NMDA receptors, which increases
intracellular cal-cium, results in the generation of nitric oxide. Although a
physiologic role for nitric oxide has been clearly established for vascular
smooth muscle, its role in synaptic transmission and synaptic plasticity
remains controversial. Perhaps the strongest case for a role of nitric oxide in
neuronal signaling in the CNS is for long-term depression of synaptic
transmission in the cerebellum.
Endocannabinoids
The
primary psychoactive ingredient in cannabis, 9-tetrahydrocannabinol
( 9-THC), affects the brain mainly by activating a specific
cannabinoid receptor, CB1. CB1 receptors are expressed at
high levels in many brain regions, and they are primarily located on
presynaptic terminals. Several endogenous brain lipids, including anandamide
and 2-arachidonylglycerol (2-AG), have been identified as CB1
ligands. These ligands are not stored, as are classic neurotransmitters, but
instead are rapidly synthesized by neurons in response to depolarization and
consequent calcium influx. Activation of metabotropic receptors (eg, by
acetylcholine and glutamate) can also activate the formation of 2-AG. In
further contradistinction to classic neurotransmitters, endogenous cannabinoids
can function as retrograde synaptic messengers: They are released from
post-synaptic neurons and travel backward across synapses, activat-ing CB1
receptors on presynaptic neurons and suppressing transmitter release. This
suppression can be transient or long lasting, depending on the pattern of
activity. Cannabinoids may affect memory, cognition, and pain perception by
this mechanism.
Related Topics