Chapter: Basic & Clinical Pharmacology : Introduction to Toxicology: Occupational & Environmental
Air Pollutants - Specific Chemicals
SPECIFIC CHEMICALS
AIR POLLUTANTS
Five
major substances account for about 98% of air pollution: carbon monoxide (CO,
about 52%), sulfur oxides (about 14%), hydrocarbons (about 14%), nitrogen
oxides (about14%),
and particulate matter (about 4%). The sources of these chemicals include
transportation, industry, generation of electric power, space heating, and
refuse disposal. Sulfur dioxide and smoke resulting from incomplete combustion
of coal have been associated with acute adverse effects, particu-larly among
the elderly and individuals with preexisting car-diac or respiratory disease.
Ambient air pollution has been implicated as a contributing factor in
bronchitis, obstructive ventilatory disease, pulmonary emphysema, bronchial
asthma, and lung cancer. EPA standards for these substances apply to the
general environment, and OSHA standards apply to workplace exposure.
Carbon Monoxide
Carbon monoxide (CO)
is a colorless, tasteless, odorless, and nonirritating gas, a byproduct of
incomplete combustion. The average concentration of CO in the atmosphere is
about 0.1 ppm; in heavy traffic, the concentration may exceed 100 ppm. The
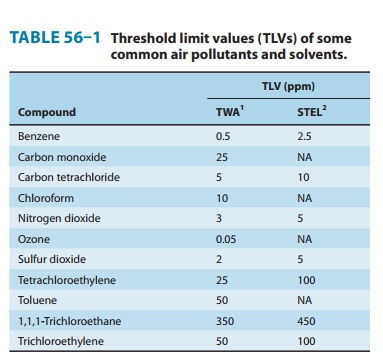
recommended 2008 threshold limit values (TLV-TWA and TLV-STEL) are shown in
Table 56–1.
A. Mechanism of Action
CO combines reversibly
with the oxygen-binding sites of hemo-globin and has an affinity for hemoglobin
that is about 220 times that of oxygen. The product formed—carboxyhemoglobin—
cannot transport oxygen. Furthermore, the presence of carboxyhe-moglobin
interferes with the dissociation of oxygen from the remaining oxyhemoglobin,
thus reducing the transfer of oxygen to tissues. The brain and the heart are
the organs most affected. Normal nonsmoking adults have carboxyhemoglobin
levels of less than 1% saturation (1% of total hemoglobin is in the form of
carboxyhemoglobin); this level has been attributed to the endog-enous formation
of CO from heme catabolism. Smokers may exhibit 5–10% saturation, depending on
their smoking habits. A person breathing air containing 0.1% CO (1000 ppm)
would have a carboxyhemoglobin level of about 50%.
B. Clinical Effects
The principal signs of
CO intoxication are those of hypoxia and progress in the following sequence:
(1) psychomotor impairment;headache and tightness in the
temporal area; (3) confusion and loss of visual acuity; (4) tachycardia,
tachypnea, syncope, and coma; and (5) deep coma, convulsions, shock, and
respiratory failure. There is great variability in individual responses to a
given carboxy-hemoglobin concentration. Carboxyhemoglobin levels below 15% may
produce headache and malaise; at 25% many workers com-plain of headache,
fatigue, decreased attention span, and loss of fine motor coordination.
Collapse and syncope may appear at around 40%; with levels above 60%, death may
ensue as a result of irrevers-ible damage to the brain and myocardium. The
clinical effects may be aggravated by heavy labor, high altitudes, and high
ambient temperatures. Although CO intoxication is usually thought of as a form
of acute toxicity, there is some evidence that chronic exposure to low levels
may lead to undesirable effects, including the develop-ment of atherosclerotic
coronary disease in cigarette smokers. The fetus may be quite susceptible to
the effects of CO exposure.
C. Treatment
In cases of acute
intoxication, removal of the individual from the exposure source and
maintenance of respiration are essential, fol-lowed by administration of
oxygen—the specific antagonist to CO—within the limits of oxygen toxicity. With
room air at 1 atm, the elimination half-time of CO is about 320 minutes; with
100% oxygen, the half-time is about 80 minutes; and with hyperbaric oxygen (2–3
atm), the half-time can be reduced to about 20 min-utes. If a hyperbaric oxygen
chamber is readily available, it should be used in the treatment of CO
poisoning for severely poisoned patients; however, there remain questions about
its effectiveness. Progressive recovery from effectively treated CO poisoning,
even of a severe degree, is often complete, although some patients dem-onstrate
persistent impairment for a prolonged period of time.
Sulfur Dioxide
Sulfur dioxide (SO2) is a colorless,
irritant gas generated primarily by the combustion of sulfur-containing fossil
fuels. The 2008 TLVs are given in Table 56–1.
A. Mechanism of Action
On
contact with moist membranes, SO2
forms sulfurous acid, which is responsible for its severe irritant effects on
the eyes, mucous mem-branes, and skin. Approximately 90% of inhaled SO2
is absorbed in the upper respiratory tract, the site of its principal effect.
The inhala-tion of SO2 causes
bronchial constriction; parasympathetic reflexes and altered smooth muscle tone
appear to be involved. Exposure to 5 ppm SO2
for 10 minutes leads to increased resistance to airflow in
Exposures of 5–10 ppm are reported to cause severe bronchospasm; 10–20%
of the healthy young adult population is estimated to be reactive to even lower
concentrations. The phenom-enon of adaptation to irritating concentrations has
been reported in workers. However, current studies have not confirmed this
phenom-enon. Asthmatic individuals are especially sensitive to SO2.
B. Clinical Effects and Treatment
The signs and symptoms
of intoxication include irritation of the eyes, nose, and throat and reflex
bronchoconstriction. In asth-matic subjects, exposure to SO2 may result in an
acute asthmatic episode. If severe exposure has occurred, delayed-onset
pulmonary edema may be observed. Cumulative effects from chronic low-level
exposure to SO2 are not striking,
particularly in humans but these effects have been associated with aggravation
of chronic cardiopulmonary disease. When combined exposure to high respi-rable
particulate loads and SO2 occurs, the mixed irritant load may increase the toxic
respiratory response. Treatment is not spe-cific for SO2 but depends on
therapeutic maneuvers used in the treatment of irritation of the respiratory
tract and asthma.
Nitrogen Oxides
Nitrogen
dioxide (NO2) is a brownish irritant gas
sometimes asso-ciated with fires. It is formed also from fresh silage; exposure
of farmers to NO2 in the
confines of a silo can lead to silo-filler’s disease. The 2008 TLVs are shown
in Table 56–1.
A. Mechanism of Action
NO2 is a relatively
insoluble deep lung irritant capable of produc-ing pulmonary edema. The type I
cells of the alveoli appear to be the cells chiefly affected on acute exposure.
At higher exposure, both type I and type II alveolar cells are damaged.
Exposure to 25 ppm of NO2 is irritating to some individuals; 50 ppm is mod-erately
irritating to the eyes and nose. Exposure for 1 hour to 50 ppm can cause
pulmonary edema and perhaps subacute or chronic pulmonary lesions; 100 ppm can
cause pulmonary edema and death.
B. Clinical Effects and Treatment
The signs and symptoms
of acute exposure to NO2 include irrita-tion of the eyes and nose, cough, mucoid or
frothy sputum produc-tion, dyspnea, and chest pain. Pulmonary edema may appear
within 1–2 hours. In some individuals, the clinical signs may sub-side in about
2 weeks; the patient may then pass into a second stage of abruptly increasing
severity, including recurring pulmonary edema and fibrotic destruction of
terminal bronchioles (bronchi-olitis obliterans). Chronic exposure of
laboratory animals to 10–25 ppm NO2 has resulted in emphysematous changes; thus,
chronic effects in humans are of concern. There is no specific treatment for
acute intoxication by NO2; therapeutic measures for the manage-ment of deep lung
irritation and noncardiogenic pulmonary edema are used. These measures include
maintenance of gas exchange with adequate oxygenation and alveolar ventilation.
Drug therapy may include bronchodilators, sedatives, and antibiotics.
Ozone
Ozone (O3) is a bluish irritant gas that occurs normally in the earth’s atmosphere, where it is an important absorbent of ultravio-let light. In the workplace, it can occur around high-voltage elec-trical equipment and around ozone-producing devices used for air and water purification. It is also an important oxidant found in polluted urban air. There is a near-linear gradient between expo-sure (1-hour level, 20–100 ppb) and response. See Table 56–1 for 2008 TLVs.
A. Clinical Effects and Treatment
O3
is an irritant of mucous membranes. Mild exposure produces upper respiratory
tract irritation. Severe exposure can cause deep lung irritation, with
pulmonary edema when inhaled at sufficient concentrations. Ozone penetration in
the lung depends on tidal volume; consequently, exercise can increase the
amount of ozone reaching the distal lung. Some of the effects of O3
resemble those seen with radiation, suggesting that O3
toxicity may result from the formation of reactive free radicals. The gas
causes shallow, rapid breathing and a decrease in pulmonary compliance.
Enhanced sensitivity of the lung to bronchoconstrictors is also observed.
Exposure around 0.1
ppm O3 for 10–30 minutes
causes irrita-tion and dryness of the throat; above 0.1 ppm, one finds changes
in visual acuity, substernal pain, and dyspnea. Pulmonary function is impaired
at concentrations exceeding 0.8 ppm. Airway hyper-responsiveness and airway
inflammation have been observed in humans.
The
response of the lung to O3 is a
dynamic one. The mor-phologic and biochemical changes are the result of both
direct injury and secondary responses to the initial damage. Long-term exposure
in animals results in morphologic and functional pul-monary changes. Chronic
bronchitis, bronchiolitis, fibrosis, and emphysematous changes have been
reported in a variety of spe-cies, including humans, exposed to concentrations
above 1 ppm. There is no specific treatment for acute O3
intoxication. Management depends on therapeutic measures used for deep lung
irritation and noncardiogenic pulmonary edema (see Nitrogen Oxides, above).
Related Topics