Chapter: Basic & Clinical Pharmacology : Introduction
Pharmacodynamic Principles
Pharmacodynamic Principles
Most
drugs must bind to a receptor to bring about an effect. However, at the
cellular level, drug binding is only the first in what is often a complex
sequence of steps:
Drug (D) + receptor-effector (R) →
drug-receptor-effector complex → effect
· D + R → drug-receptor complex → effector molecule → effect
·
D +
R →
D-R complex →
activation of coupling molecule → effector molecule → effect
·
Inhibition of metabolism of endogenous activator → increased activator
action on an effector molecule → increased effect
Note
that the final change in function is accomplished by an effector mechanism. The effector may be part of the
receptormolecule or may be a separate molecule. A very large number of
receptors communicate with their effectors through coupling mol-ecules.
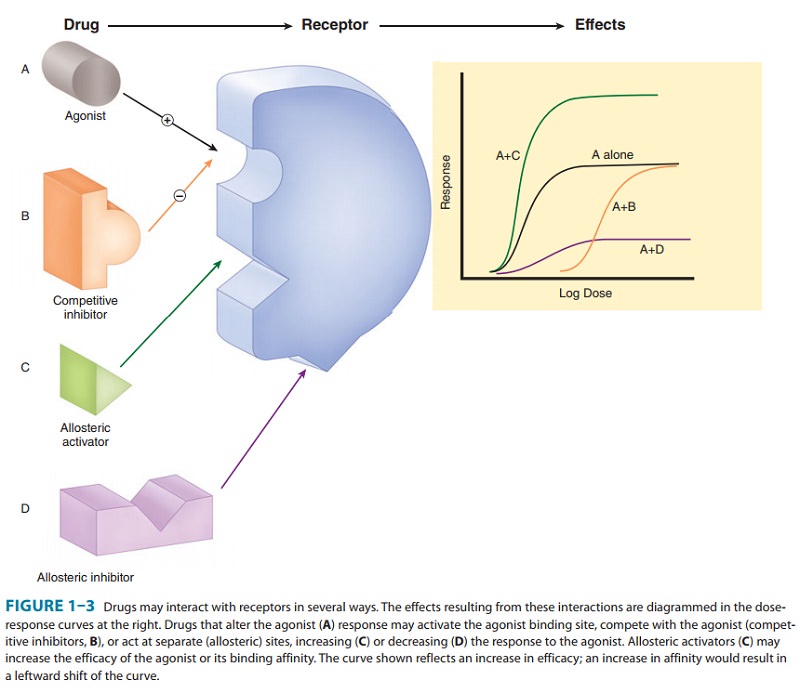
A. Types of Drug-Receptor Interactions
Agonist drugs bind to andactivatethe receptor in some fashion,which directly or indirectly
brings about the effect (Figure 1–3A). Receptor activation involves a change in
conformation in the cases that have been studied at the molecular structure
level. Some receptors incorporate effector machinery in the same mol-ecule, so
that drug binding brings about the effect directly, eg, opening of an ion
channel or activation of enzyme activity. Other receptors are linked through
one or more intervening coupling molecules to a separate effector molecule. The
five major types of drug-receptor-effector coupling systems are discussed later.
Pharmacologic antagonist drugs, by
bind-ing to a receptor, compete with and prevent binding by other molecules.
For example, acetylcholine receptor blockers such as atropine are antagonists
because they prevent access of acetylcho-line and similar agonist drugs to the acetylcholine
receptor site and they stabilize the receptor in its inactive state (or some
state other than the acetylcholine-activated state). These agents reduce the
effects of acetylcholine and similar molecules in the body (Figure 1–3B), but
their action can be overcome by increasing the dosage of agonist. Some
antagonists bind very tightly to the recep-tor site in an irreversible or
pseudoirreversible fashion and cannot be displaced by increasing the agonist
concentration. Drugs that bind to the same receptor molecule but do not prevent
binding of the agonist are said to act allosterically
and may enhance (Figure 1–3C) or inhibit (Figure 1–3D) the action of the
agonist mole-cule. Allosteric inhibition is not overcome by increasing the dose
of agonist.
B. Agonists That Inhibit Their
Binding Molecules
Some
drugs mimic agonist drugs by inhibiting the molecules responsible for
terminating the action of an endogenous agonist. For example,
acetylcholinesterase inhibitors, by
slowing the destruction of endogenous acetylcholine, cause cholinomimetic
effects that closely resemble the actions of cholinoceptor agonist molecules even though cholinesterase inhibitors do not bind
or only incidentally bind to cholinoceptors . Because they amplify the effects
of physiologically released agonist ligands, their effects are sometimes more
selective and less toxic than those of exogenous agonists.
C. Agonists, Partial Agonists, and Inverse Agonists
Figure
1–4 describes a useful model of drug-receptor interaction. As indicated, the
receptor is postulated to exist in the inactive,
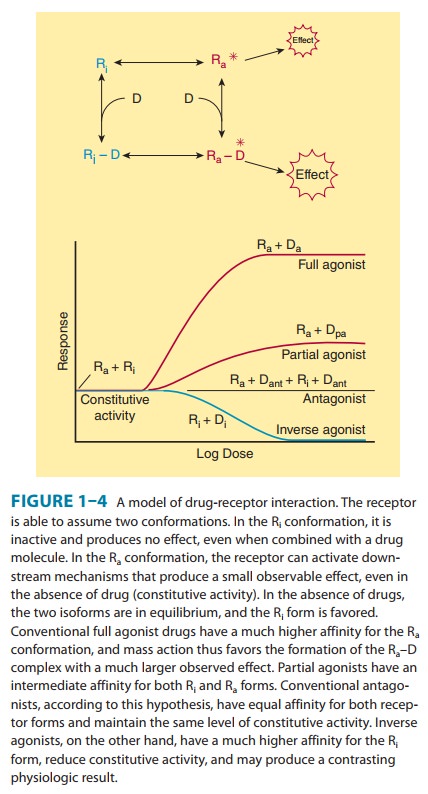
Thermodynamic considerations indicate that even in the absence of any agonist,
some of the receptor pool must exist in the Ra form some of the time
and may produce the same physiologic effect as agonist-induced activity. This
effect, occurring in the absence of agonist, is termed constitutive activity. Agonists are those drugs that have a much
higher affinity for the Ra configuration and stabilize it, so that a
large percentage of the total pool resides in the Ra–D fraction and
a large effect is produced. The recognition of constitutive activity may depend
on the receptor density, the concentration of coupling molecules (if a coupled
system), and the number of effectors in the system.
Many
agonist drugs, when administered at concentrations suf-ficient to saturate the
receptor pool, can activate their receptor-ef-fector systems to the maximum extent
of which the system is capable; that is, they cause a shift of almost all of
the receptor pool to the Ra–D pool. Such drugs are termed full agonists. Other drugs, called partial agonists, bind to the same
receptors and activate themin the same way but do not evoke as great a
response, no matter how high the concentration. In the model in Figure 1–4,
partial agonists do not stabilize the Ra configuration as fully as
full ago-nists, so that a significant fraction of receptors exists in the Ri–D
pool. Such drugs are said to have low intrinsic
efficacy. Thus, pindolol, a β-adrenoceptor partial agonist, may act either
as an agonist (if no full agonist is present) or as an antagonist (if a full
agonist such as epinephrine is present). Intrinsic efficacy is independent of
affinity (as usually measured) for the receptor.In the same model, conventional
antagonist action can be explained as fixing the fractions of drug-bound Ri
and Ra in the same relative amounts as in the absence of any drug.
In this situa-tion, no change will be observed, so the drug will appear to be
without effect. However, the presence of the antagonist at the receptor site
will block access of agonists to the receptor and pre-vent the usual agonist
effect. Such blocking action can be termed neutral
antagonism.
What
will happen if a drug has a much stronger affinity for the Ri than
for the Ra state and stabilizes a large fraction in the Ri–D
pool? In this scenario the drug would reduce any constitutive activity, thus
resulting in effects that are the opposite of the effects produced by
conventional agonists at that receptor. Such drugs have been termed inverse agonists (Figure 1–4). One of
the best documented examples of such a system is the γ-aminobutyric acid (GABAA)
receptor-effector (a chloride channel) in the nervous system. This receptor is
activated by the endogenous transmitter GABA and causes inhibition of
postsynaptic cells. Conventional exogenous agonists such as benzodiazepines also
facilitate the receptor-effector system and cause GABA-like inhibition with
sedation as the therapeutic result. This inhibition can be blocked by
conventional neutral antagonists such as flumazenil. In addi-tion, inverse
agonists have been found that cause anxiety and agitation, the inverse of
sedation . Similar inverse agonists have been found for β-adrenoceptors,
histamine H1 and H2 receptors, and several other receptor
systems.
D. Duration of Drug Action
Termination
of drug action is a result of one of several processes. In some cases, the
effect lasts only as long as the drug occupies the receptor, and dissociation
of drug from the receptor automatically terminates the effect. In many cases,
however, the action may persist after the drug has dissociated because, for
example, some coupling molecule is still present in activated form. In the case
of drugs that bind covalently to the receptor site, the effect may per-sist
until the drug-receptor complex is destroyed and new recep-tors or enzymes are
synthesized, as described previously for aspirin. In addition, many
receptor-effector systems incorporate desensiti-zation mechanisms for
preventing excessive activation when ago-nist molecules continue to be present
for long periods.
E. Receptors and Inert Binding Sites
To
function as a receptor, an endogenous molecule must first be selective in choosing ligands (drug
molecules) to bind; and second,it must change
its function upon binding in such a way that the function of the biologic
system (cell, tissue, etc) is altered. The selectivity characteristic is
required to avoid constant activation of the receptor by promiscuous binding of
many different ligands. The ability to change function is clearly necessary if
the ligand is to cause a pharmacologic effect. The body contains a vast array
of molecules that are capable of binding drugs, however, and not all of these
endogenous molecules are regulatory molecules. Binding of a drug to a
nonregulatory molecule such as plasma albumin will result in no detectable
change in the function of the biologic system, so this endogenous molecule can
be called an inert binding site.
Such binding is not completely without significance, however, because it
affects the distribution of drug within the body and determines the amount of
free drug in the circulation. Both of these factors are of pharmacokinetic
importance.
Related Topics