Chapter: Basic & Clinical Pharmacology : Introduction
Pharmacokinetic Principles
Pharmacokinetic Principles
In practical therapeutics, a drug should be able to reach its intended site of action after administration by some convenient route. In many cases, the active drug molecule is sufficiently lipid-soluble and stable to be given as such. In some cases, however, an inactive precursor chemical that is readily absorbed and distrib-uted must be administered and then converted to the active drug by biologic processes—inside the body. Such a precursor chemical is called a prodrug.In only a few situations is it possible to apply a drug directly to its target tissue, eg, by topical application of an anti-inflammatory agent to inflamed skin or mucous membrane. Most often, a drug is administered into one body compartment, eg, the gut, and must move to its site of action in another compartment, eg, the brain in the case of an antiseizure medication. This requires that the drug be absorbed into the blood from its site of administration and distributed to its site of action, permeating through the various barriers that separate these compartments.
For a drug given orally to produce an effect
in the central nervous system, these barriers include the tissues that make up
the wall of the intestine, the walls of the capillaries that perfuse the gut,
and the blood-brain barrier, the walls of the capillaries that perfuse the
brain. Finally, after bringing about its effect, a drug should be eliminated at a reason-able rate by
metabolic inactivation, by excretion from the body, or by a combination of
these processes.
A. Permeation
Drug
permeation proceeds by several mechanisms. Passive diffu-sion in an aqueous or
lipid medium is common, but active pro-cesses play a role in the movement of
many drugs, especially those whose molecules are too large to diffuse readily
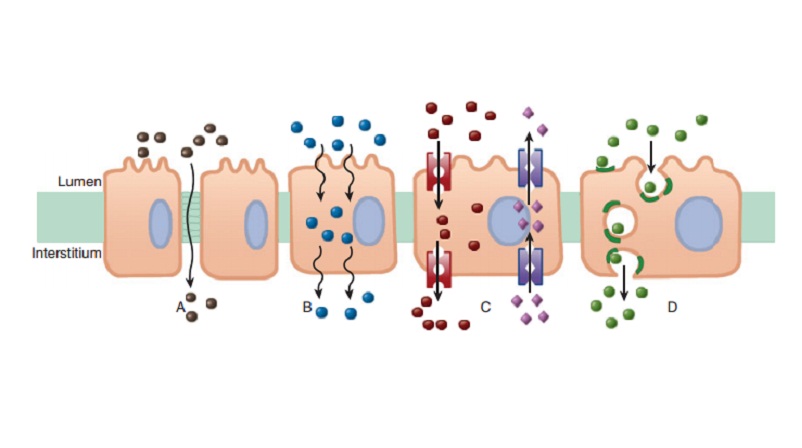
(Figure 1–5).
1. Aqueous diffusion—Aqueous diffusion occurs within thelarger aqueous compartments of the body (interstitial space, cyto-sol, etc) and across epithelial membrane tight junctions and the endothelial lining of blood vessels through aqueous pores that—in some tissues—permit the passage of molecules as large as MW 20,000–30,000.* See Figure 1–5A.
Aqueous
diffusion of drug molecules is usually driven by the concentration gradient of
the permeating drug, a downhill move-ment described by Fick’s law . Drug
molecules that are bound to large plasma proteins (eg, albumin) do not permeate
most vascular aqueous pores. If the drug is charged, its flux is also
influenced by electrical fields (eg, the membrane potential and—in parts of the
nephron—the transtubular potential).
2.
Lipid diffusion—Lipid diffusion is the
most important limit-ing factor for drug permeation because of the large number of lipid barriers that separate the
compartments of the body. Because these lipid barriers separate aqueous compartments, thelipid:aqueous partition coefficient of a drug determines howreadily
the molecule moves between aqueous and lipid media. In the case of weak acids
and weak bases (which gain or lose electrical charge-bearing protons, depending
on the pH), the ability to move from aqueous to lipid or vice versa varies with
the pH of the medium, because charged molecules attract water molecules. The
ratio of lipid-soluble form to water-soluble form for a weak acid or weak base
is expressed by the Henderson-Hasselbalch equation (described in the following
text). See Figure 1–5B.
3. Special carriers—Special carrier
molecules exist for manysubstances that are important for cell function and too
large or too insoluble in lipid to diffuse passively through membranes, eg,
peptides, amino acids, and glucose. These carriers bring about movement by
active transport or facilitated diffusion and, unlike passive diffusion, are
selective, saturable, and inhibitable. Because many drugs are or resemble such
naturally occurring peptides, amino acids, or sugars, they can use these
carriers to cross mem-branes. See Figure 1–5C.
Many
cells also contain less selective membrane carriers that are specialized for
expelling foreign molecules. One large family of such transporters binds
adenosine triphosphate (ATP) and is called the ABC (ATP-binding cassette)
family. This family includes the P-glycoprotein
or multidrug resistance type 1
(MDR1) trans-porter found in the brain, testes, and other tissues, and in somedrug-resistant
neoplastic cells, Table 1–2. Similar transport molecules from the ABC family,
the multidrug resistance-associated
pro-tein (MRP) transporters, play important roles in the excretion ofsome
drugs or their metabolites into urine and bile and in the resistance of some
tumors to chemotherapeutic drugs. Several other transporter families have been
identified that do not bind ATP but use ion gradients to drive transport. Some
of these (the solute car-rier [SLC] family) are particularly important in the
uptake of neurotransmitters across nerve-ending membranes. The latter car-riers
are discussed in more detail later.
4. Endocytosis and exocytosis—A few substances are
so large orimpermeant that they can enter cells only by endocytosis, the
pro-cess by which the substance is bound at a cell-surface receptor,
engulfed
by the cell membrane, and carried into the cell by pinching off of the newly
formed vesicle inside the membrane. The substance can then be released inside
the cytosol by breakdown of the vesicle membrane, Figure 1–5D. This process is
responsible for the trans-port of vitamin B12, complexed with a
binding protein (intrinsic factor) across the wall of the gut into the blood.
Similarly, iron is transported into hemoglobin-synthesizing red blood cell
precursors in association with the protein transferrin. Specific receptors for
the transport proteins must be present for this process to work.
The
reverse process (exocytosis) is responsible for the secretion of many
substances from cells. For example, many neurotransmit-ter substances are
stored in membrane-bound vesicles in nerve endings to protect them from
metabolic destruction in the cyto-plasm. Appropriate activation of the nerve
ending causes fusion of the storage vesicle with the cell membrane and
expulsion of its contents into the extracellular space .
B. Fick’s Law of Diffusion
The
passive flux of molecules down a concentration gradient is given by Fick’s law:

where
C1 is the higher concentration, C2 is the lower
concentra-tion, area is the cross-sectional area of the diffusion path,
perme-ability coefficient is a measure of the mobility of the drug molecules in
the medium of the diffusion path, and thickness is the thickness (length) of
the diffusion path. In the case of lipid diffusion, the lipid:aqueous partition
coefficient is a major deter-minant of mobility of the drug, because it
determines how readily the drug enters the lipid membrane from the aqueous
medium.
C. Ionization of Weak Acids and
Weak Bases; the Henderson-Hasselbalch Equation
The
electrostatic charge of an ionized molecule attracts water dipoles and results
in a polar, relatively water-soluble and lipid-in-soluble complex. Because
lipid diffusion depends on relatively high lipid solubility, ionization of
drugs may markedly reduce their abil-ity to permeate membranes. A very large
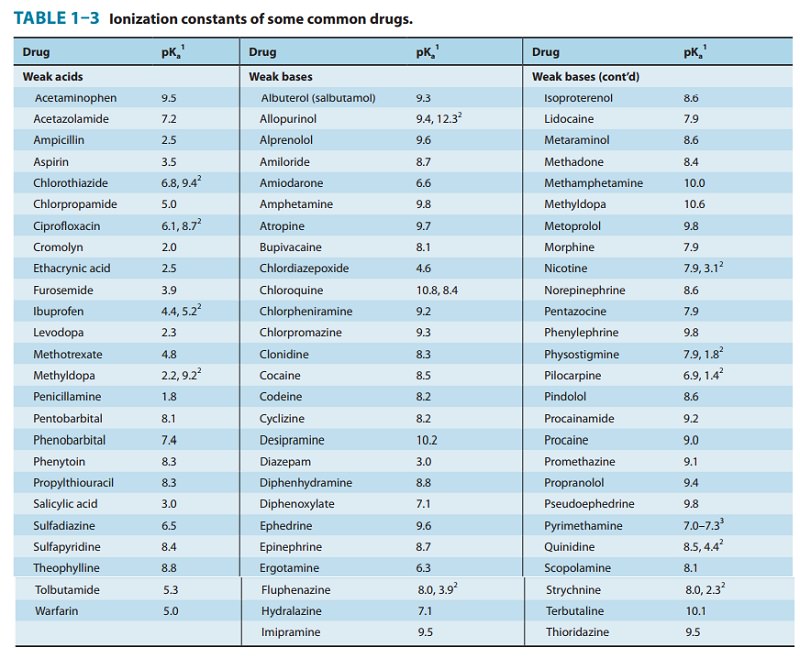
percentage of the drugs in use are weak acids or weak bases (Table 1–3). For
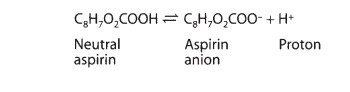
drugs, a weak acid is best defined as a neutral molecule that can reversibly
dis-sociate into an anion (a negatively charged molecule) and a proton (a
hydrogen ion). For example, aspirin dissociates as follows:
A
drug that is a weak base can be defined as a neutral molecule that can form a
cation (a positively charged molecule) by combin-ing with a proton. For
example, pyrimethamine, an antimalarial drug, undergoes the following
association-dissociation process:
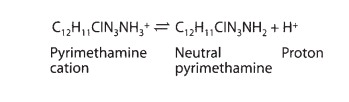
Note
that the protonated form of a weak acid is the neutral, more lipid-soluble
form, whereas the unprotonated form of a weak base is the neutral form. The law
of mass action requires that these reactions move to the left in an acid environment
(low pH, excess protons available) and to the right in an alkaline
environ-ment. The Henderson-Hasselbalch equation relates the ratio of
protonated to unprotonated weak acid or weak base to the mole-cule’s pKa
and the pH of the medium as follows:
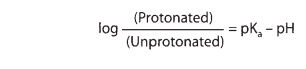
This
equation applies to both acidic and basic drugs. Inspection confirms that the
lower the pH relative to the pKa, the greater will be the fraction
of drug in the protonated form. Because the uncharged form is the more
lipid-soluble, more of a weak acid will be in the lipid-soluble form at acid
pH, whereas more of a basic drug will be in the lipid-soluble form at alkaline
pH.
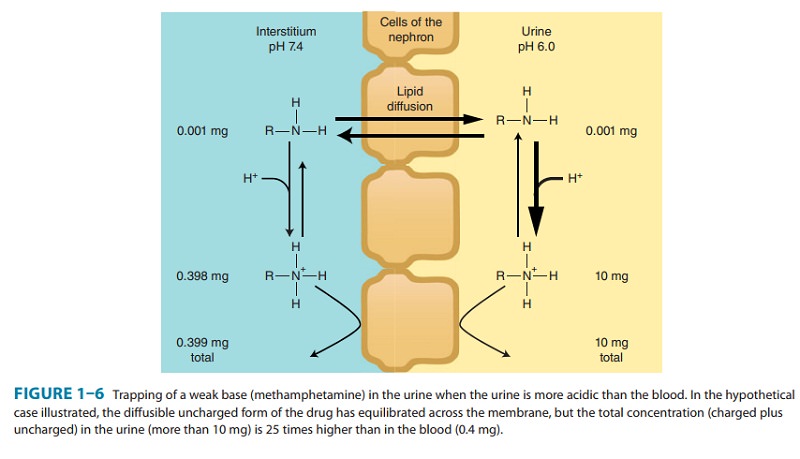
Application
of this principle is made in the manipulation of drug excretion by the kidney.
Almost all drugs are filtered at the glomerulus. If a drug is in a
lipid-soluble form during its passage down the renal tubule, a significant
fraction will be reabsorbed by simple passive diffusion. If the goal is to
accelerate excretion of the drug (eg, in a case of drug overdose), it is
important to pre-vent its reabsorption from the tubule. This can often be
accom-plished by adjusting urine pH to make certain that most of the drug is in
the ionized state, as shown in Figure 1–6. As a result of this partitioning
effect, the drug is “trapped” in the urine. Thus, weak acids are usually
excreted faster in alkaline urine; weak bases are usually excreted faster in
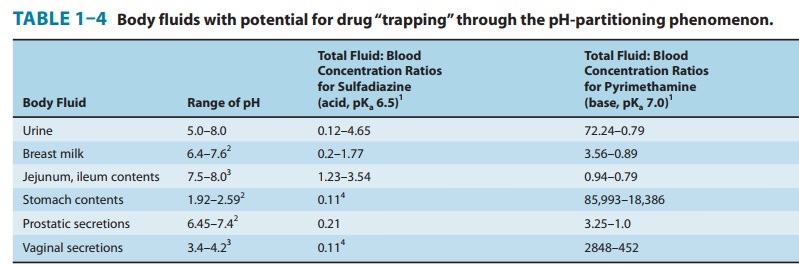
acidic urine. Other body fluids in which pH differences from blood pH may cause
trapping or reabsorption are the contents of the stomach and small intestine;
breast
milk; aqueous humor; and vaginal and prostatic secretions (Table 1–4).
As
suggested by Table 1–3, a large number of drugs are weak bases. Most of these
bases are amine-containing molecules. The nitrogen of a neutral amine has three
atoms associated with it plus a pair of unshared electrons (see the display
that follows). The three atoms may consist of one carbon (designated “R”) and
two hydrogens (a primary amine), two
carbons and one hydrogen (a secondary
amine), or three carbon atoms (a
tertiary amine).Each of these three forms may reversibly bind a proton with
the unshared electrons. Some drugs have a fourth carbon-nitrogen bond; these
are quaternary amines. However, the
quaternary amine is permanently charged and has no unshared electrons with
which to reversibly bind a proton. Therefore, primary, secondary, and tertiary
amines may undergo reversible protonation and vary their lipid solubility with
pH, but quaternary amines are always in the poorly lipid-soluble charged form.
Related Topics