Chapter: Modern Analytical Chemistry: Equilibrium Chemistry
Activity Effects
Activity Effects
Suppose you need to prepare
a buffer with a pH of 9.36.
Using the Henderson– Hasselbalch equation, you calculate the amounts of acetic acid and sodium
acetate needed and prepare
the buffer. When you measure
the pH, however,
you find that it
is 9.25. If you have
been careful in your calculations and measurements, what
can account for the difference between
the obtained and expected pHs? In this section,
we will examine an important limitation to our use of equilibrium constants and learn how this limitation can be corrected.
Careful measurements of the solubility of AgIO3 show
that it increases in the presence of KNO3, even
though neither K+ or NO3–
participates in the
solubility reaction.
Clearly the equilibrium position for the reaction
AgIO3(s) < == === > Ag+(aq)+ IO3–(aq)
depends on the composition of the solution.
When the solubility product for AgIO3 is calculated using the
equilibrium concentrations of Ag+ and IO3–
Ksp = [Ag+][IO3–]
its apparent value increases when an inert electrolyte such as
KNO3 is added.
Why should adding
an inert electrolyte affect the equilibrium position of a chemical reaction? We can explain the effect of KNO3 on the solubility of AgIO3 by considering the reaction on a microscopic scale. The solution
in which equilibrium is established contains a variety of cations and
anions—K+, Ag+,
H3O+, NO3–,
IO3– and OH–.
Although the solution is homogeneous, on the average, there are more anions in regions near Ag+ ions, and more cations
in regions near IO3– ions. Thus, Ag+ and IO3– are surrounded by charged
ionic atmospheres that partially screen
the ions from each
other. The formation of AgIO3 requires
the disruption of the ionic atmospheres surrounding the Ag+ and IO3– ions. Increasing the concentrations
of ions in solution, by adding
KNO3, increases the size of these ionic
atmospheres. Since more energy
is now required to disrupt
the ionic atmospheres, there is a decrease in the formation of AgIO3, and an apparent
increase in the equilibrium
constant.
The ionic composition of a solution frequently is expressed by
its ionic strength, ÎĽ
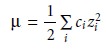
where ci
and zi are the
concentration and charge of the ith
ion.
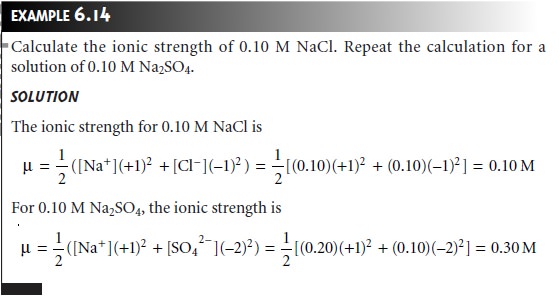
Note that the
unit for ionic
strength is molarity, but that the
molar ionic strength need not match the
molar concentration of the electrolyte. For a 1:1
electrolyte, such as NaCl, ionic strength
and molar concentration are identical.
The ionic strength of a 2:1 electrolyte, such as Na2SO4, is three times
larger than the elec-
trolyte’s molar concentration.
The
true
thermodynamic equilibrium constant
is
a function
of activity rather than concentration. The activity of a species,
aA, is defined as the prod-
uct of its molar concentration, [A], and a solution-dependent activity coeffi- cient, ÎłA.
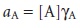
The true thermodynamic equilibrium constant, Ksp, for the solubility of AgIO3,
therefore, is
Ksp = (aAg+)(aIO3
–) = [Ag+][IO3-](γAg+)(γIO3–)
To accurately calculate the solubility of AgIO3, we must know the activity
coeffi- cients for Ag+ and IO3 –.
For gases, pure solids, pure liquids, and nonionic solutes,
activity coefficients are approximately unity under most reasonable experimental conditions. For reac- tions involving only these species,
differences between activity and concentration are negligible. Activity coefficients for ionic solutes,
however, depend on the ionic composition of the solution. It is possible, using the extended
Debye–Hückel the- ory,* to calculate activity coefficients using equation 6.50
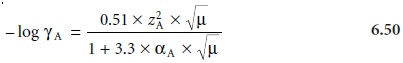
where ZA is the charge
of the ion, αA is the effective diameter of the hydrated ion in
nanometers (Table 6.1), μ is the solution’s ionic strength, and 0.51 and 3.3 are con-
stants appropriate for aqueous solutions at 25 °C.
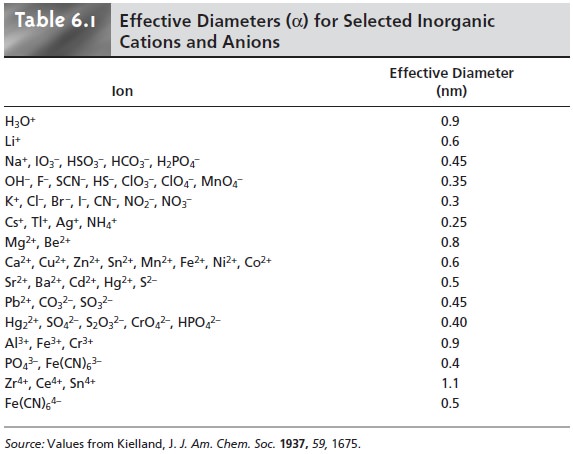
Several features of equation 6.50 deserve mention. First, as the ionic strength approaches zero, the activity coefficient approaches a value of one. Thus, in a solu- tion where the ionic strength is zero, an ion’s activity and concentration are identi- cal. We can take advantage of this fact to determine a reaction’s thermodynamic equilibrium constant. The equilibrium constant based on concentrations is mea- sured for several increasingly smaller ionic strengths and the results extrapolated back to zero ionic strength to give the thermodynamic equilibrium constant.
Second, activity coefficients are smaller, and thus activity
effects are more important,
for ions with higher charges
and smaller effective
diameters. Finally, the extended
Debye–Hückel equation provides reasonable activity coefficients
for ionic strengths of less than 0.1. Modifications to the extended
Debye–Hückel equation, which extend
the calculation of activity coefficients to higher ionic
strength, have been proposed.

As this example
shows, failing to correct for the effect of ionic strength can lead
to significant differences between calculated and actual concentrations. Neverthe- less, it is not unusual
to ignore activities and assume that the equilibrium constant is expressed in terms of concentrations. There is a practical reason for this—in
an analysis one rarely
knows the composition, much less the
ionic strength of a sample solution. Equilibrium calculations are often used as a guide when developing an an-
alytical method. Only by conducting the analysis and evaluating the results can we
judge whether our theory matches
reality.
Related Topics