Chapter: Basic & Clinical Pharmacology : Antiseizure Drugs
Valproic Acid & Sodium Valproate
VALPROIC ACID & SODIUM
VALPROATE
Sodium
valproate, also used as the free acid, valproic acid, was found to have
antiseizure properties when used as a solvent in the search for other drugs
effective against seizures. It was marketed in France in 1969 but was not
licensed in the USA until 1978. Valproic acid is fully ionized at body pH, and
for that reason the active form of the drug may be assumed to be the valproate
ion regardless of whether valproic acid or a salt of the acid is administered.
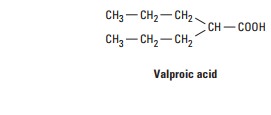
Chemistry
Valproic
acid is one of a series of fatty carboxylic acids that have antiseizure
activity; this activity appears to be greatest for carbon chain lengths of five
to eight atoms. The amides and esters of valproic acid are also active
antiseizure agents.
Mechanism of Action
The
time course of valproate’s anticonvulsant activity appears to be poorly
correlated with blood or tissue levels of the parent drug, an observation
giving rise to considerable speculation regarding both the active species and
the mechanism of action of valproic acid. Valproate is active against both
pentylenetetrazol and maxi-mal electroshock seizures. Like phenytoin and
carbamazepine, valproate blocks sustained high-frequency repetitive firing of
neu-rons in culture at therapeutically relevant concentrations. Its action
against partial seizures may be a consequence of this effect on Na+
currents. Blockade of NMDA receptor-mediated excita-tion may also be important.
Much attention has been paid to the effects of valproate on GABA. Several
studies have shown increased levels of GABA in the brain after administration
of val-proate, although the mechanism for this increase remains unclear. An
effect of valproate to facilitate glutamic acid decarboxylase (GAD), the enzyme
responsible for GABA synthesis, has been described. An inhibitory effect on the
GABA transporter GAT-1 may contribute. At very high concentrations, valproate
inhibits GABA transaminase in the brain, thus blocking degradation of GABA.
However, at the relatively low doses of valproate needed to abolish
pentylenetetrazol seizures, brain GABA levels may remain unchanged. Valproate
produces a reduction in the aspartate con-tent of rodent brain, but the
relevance of this effect to its anticon-vulsant action is not known.
Valproic
acid is a potent inhibitor of histone deacetylase and through this mechanism
changes the transcription of many genes. A similar effect, but to a lesser
degree, is shown by some other antiseizure drugs (topiramate, carbamazepine,
and a metabolite of levetiracetam).
Clinical Uses
Valproate
is very effective against absence seizures and is often preferred to
ethosuximide when the patient has concomitant generalized tonic-clonic attacks.
Valproate is unique in its ability to control certain types of myoclonic
seizures; in some cases the effect is very dramatic. The drug is effective in
tonic-clonic seizures, especially those that are primarily generalized. A few
patients with atonic attacks may also respond, and some evidence suggests that
the drug is effective in partial seizures. Its use in epilepsy is at least as
broad as that of any other drug. Intravenous formulations are occasionally used
to treat status epilepticus.Other uses of valproate include management of
bipolar disorder and migraine prophylaxis.
Pharmacokinetics
Valproate
is well absorbed after an oral dose, with bioavailability greater than 80%.
Peak blood levels are observed within 2 hours. Food may delay absorption, and
decreased toxicity may result if the drug is given after meals.
Valproic
acid is 90% bound to plasma proteins, although the fraction bound is somewhat
reduced at blood levels greater than 150 mcg/mL. Since valproate is both highly
ionized and highly protein-bound, its distribution is essentially confined to
extracellular water, with a volume of
distribution of approximately 0.15 L/kg. At higher doses, there is an increased
free fraction of valproate, resulting in lower total drug levels than expected.
It may be clinically useful, therefore, to measure both total and free drug
levels. Clearance for valproate is low and dose dependent; its half-life varies
from 9 to 18 hours. Approximately 20% of the drug is excreted as a direct
conjugate of valproate.
The
sodium salt of valproate is marketed in Europe as a tablet and is quite
hygroscopic. In Central and South America, the mag-nesium salt is available,
which is considerably less hygroscopic. The free acid of valproate was first
marketed in the USA in a cap-sule containing corn oil; the sodium salt is also
available in syrup, primarily for pediatric use. An enteric-coated tablet of
divalproex sodium is also marketed in the USA. This improved product, a 1:1
coordination compound of valproic acid and sodium valproate, is as bioavailable
as the capsule but is absorbed much more slowly and is preferred by many
patients. Peak concentrations following administration of the enteric-coated
tablets are seen in 3–4 hours. Various extended-release preparations are
available; not all are bioequivalent and may require dosage adjustment.
Therapeutic Levels & Dosage
Dosages
of 25–30 mg/kg/d may be adequate in some patients, but others may require 60
mg/kg/d or even more. Therapeutic levels of valproate range from 50 to 100
mcg/mL.
Drug Interactions
Valproate
displaces phenytoin from plasma proteins. In addition to binding interactions,
valproate inhibits the metabolism of several drugs, including phenobarbital,
phenytoin, and carbamazepine, leading to higher steady-state concentrations of
these agents. The inhibition of phenobarbital metabolism, for example, may
cause levels of the barbiturate to rise steeply, causing stupor or coma.
Valproate can dramatically decrease the clearance of lamotrigine.
Toxicity
The
most common dose-related adverse effects of valproate are nausea, vomiting, and
other gastrointestinal complaints such as abdominal pain and heartburn. The
drug should be started gradu-ally to avoid these symptoms. Sedation is uncommon
with val-proate alone but may be striking when valproate is added to
phenobarbital. A fine tremor is frequently seen at higher levels. Other
reversible adverse effects, seen in a small number of patients, include weight
gain, increased appetite, and hair loss.The idiosyncratic toxicity of valproate
is largely limited to hepatotoxicity, but this may be severe; there seems
little doubt that the hepatotoxicity of valproate has been responsible for more
than 50 fatalities in the USA alone. The risk is greatest for patients under 2
years of age and for those taking multiple medications. Initial aspartate
aminotransferase values may not be elevated in susceptible patients, although
these levels do eventually become abnormal. Most fatalities have occurred
within 4 months after initiation of therapy. Some clinicians recommend
treatment with oral or intravenous L-carnitine as soon as severe hepatotoxicity
is suspected. Careful monitoring of liver function is recommended when starting
the drug; the hepatotoxicity is reversible in some cases if the drug is
withdrawn. The other observed idiosyncratic response with valproate is
thrombocytopenia, although documented cases of abnormal bleeding are lacking.
It should be noted that valproate is an effective and popular antiseizure drug
and that only a very small number of patients have had severe toxic effects
from its use.
Several
epidemiologic studies of valproate have confirmed a substantial increase in the
incidence of spina bifida in the off-spring of women who took valproate during
pregnancy. In addi-tion, an increased incidence of cardiovascular, orofacial,
and digital abnormalities has been reported. These observations must be
strongly considered in the choice of drugs during pregnancy.
Related Topics