Chapter: Basic & Clinical Pharmacology : Antiseizure Drugs
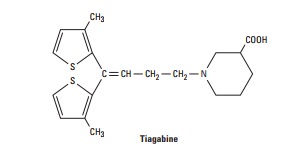
Tiagabine
TIAGABINE
Tiagabine
is a derivative of nipecotic acid and was “rationally designed” as an inhibitor
of GABA uptake (as opposed to discov-ery through random screening).
Mechanism of Action
Tiagabine
is an inhibitor of GABA uptake in both neurons and glia. It preferentially inhibits
the transporter isoform 1 (GAT-1) rather than GAT-2 or GAT-3 and increases
extracellular GABA levels in the forebrain and hippocampus where GAT-1 is
prefer-entially expressed. It prolongs the inhibitory action of synapti-cally
released GABA, but its most significant effect may be potentiation of tonic
inhibition. In rodents, it is potent against kindled seizures but weak against
the maximal electroshock model, consistent with its predominant action in the
forebrain and hippocampus.
Clinical Uses
Tiagabine
is indicated for the adjunctive treatment of partial sei-zures and is effective
in doses ranging from 16 to 56 mg/d. Divided doses as often as four times daily
are sometimes required. Minor adverse events are dose related and include
nervousness, dizziness, tremor, difficulty in concentrating, and depression.
Excessive confusion, somnolence, or ataxia may require discon-tinuation.
Psychosis occurs rarely. The drug can cause
seizures in some patients, notably those taking the drug for other indications.
Rash is an uncommon idiosyncratic adverse effect.
Pharmacokinetics
Tiagabine
is 90–100% bioavailable, has linear kinetics, and is highly protein bound. The
half-life is 5–8 hours and decreases in the presence of enzyme-inducing drugs.
Food decreases the peak plasma concentration but not the area under the
concentration curve . Hepatic impairment causes a slight decrease in clearance
and may necessitate a lower dose. The drug is oxidized in the liver by CYP3A.
Elimination is primarily in the feces (60–65%) and urine (25%).
Related Topics