Chapter: Biotechnology Applying the Genetic Revolution: Genomics and Gene Expression
Monitoring Gene Expression Using Whole-Genome Tiling Arrays
MONITORING
GENE EXPRESSION USING WHOLE-GENOME TILING ARRAYS
Whole-genome tiling arrays
(WGAs) are oligonucleotide microarrays that cover the entire genome. The first
entire genome to be represented by a whole-genome array was from Arabidopsis. A
gene chip was designed to have 25-mer oligonucleotides that overlapped each
other and covered the entire sequence of the genome. Complementary
oligonucleotide sequences were tiled back to back along each entire chromosome
and ordered so that the array could be conveniently analyzed for gene
expression (Fig. 8.23).
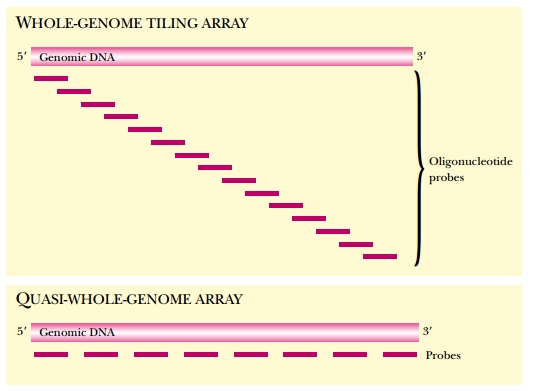
For the human genome, tiling
arrays have been made to cover the entire sequences of chromosomes 21 and 22.
These also use 25-mer oligonucleotides, but rather than being overlapped, the
oligonucleotides were spaced 35 base pairs apart along the sequence.
These are therefore strictly
only “quasi-whole-genome arrays.” Compared to arrays that include only known
genes, tiling arrays have the potential to identify novel regions that are transcribed,
whether these encode unknown protein-encoding genes or nontranslated RNA. The
RNA extracted from many different cell lines and tissues has been used to
monitor gene expression, assess differences in splicing patterns, find new
genes, and find RNA-binding protein target sequences.
The most interesting finding
from studying human chromosomes 21 and 22 is that much larger portions of these
chromosomes are transcribed into mRNA than previously predicted from computer
analyses of exon regions. About 90% of the transcribed regions occurred outside
the known exons. The majority of the transcribed regions generated noncoding
RNA, mostly of less than 75 base pairs in length. This suggests that noncoding
RNA may have a much greater role in human biology than previously thought.
These arrays have also identified new exons that were previously unknown. In
addition, these arrays can identify novel alternatively spliced proteins. The
WGAs for chromosomes 21 and 22 have also been used to compare the level of expression
of exons within the same gene. About 80% of the genes had exons with varied
levels of expression, implying most genes have some sort of alternate splicing.
Another potential use for
whole genome arrays is to analyze results of chromatin immunoprecipitation
(ChIP). ChIP begins by crosslinking all the various transcription factors to
chromatin, essentially freezing them in place. Next the chromatin is sheared
into smaller fragments, and the DNA/transcription factor complexes are
isolated. Affinity purification isolates one particular transcription factor
from all the others (e.g., antibodies to the transcription factor Jun isolates
all the Jun/DNA complexes from this mixture). Finally the DNA sequences that
are bound to the chosen transcription factor are identified using WGA.
The entire procedure,
including the analysis on a gene chip, is called ChIP-chip. This type of
analysis can precisely identify transcription factor binding sites on a variety
of genes. Curiously, binding locations for NF-κB, for example, have been found
within both coding and noncoding regions, such as introns or the 3′ ends of
genes. These surprising findings suggest that transcription factors may also
function outside of the traditional upstream promoter region.
Another use for WGA is to
identify regions of the genome that are methylated. Methylation prevents the
inappropriate expression of various genes, especially those used only during
development of young organisms, or those genes from transposons or viruses that
could be detrimental. Cancerous cells have methylation patterns much different
from those of normal cells, suggesting that this type of regulation is critical
to proper growth control of normal cells. In order to identify the methylated
regions, genomic DNA is first treated with sodium
bisulfite, which deaminates nonmethylated cytosine to uracil, yet does not
affect methylated cytosine. The treated DNA is then hybridized to a WGA. Those
regions with nonmethylated cytosine no longer hybridize to the array because
the cytosines have been converted to uracil (which pairs with A, not G). Those
regions of the genome that are methylated still hybridize well because
methylated cytosine and guanine form a stable base pair.
Of course, finding genetic
variations and polymorphisms is critical to genome analysis, and whole-genome
arrays offer a nonbiased method to analyze samples. In fact, a WGA that has the
reference sequence for the human genome can be used to identify and catalogue
all different types of polymorphisms, including SNPs, VNTRs, and repetitive
elements. In fact, an overlapping WGA made to the entire reference sequence of
the human genome spaced at a single base pair could be used to effectively
resequence the entire genome with ease and speed.
Related Topics