Chapter: Biochemical Pharmacology : G protein-coupled receptors
Cholinergic antagonists
Cholinergic antagonists
Cholinergic antagonists have
a larger therapeutic role than agonists. Again, we can distinguish drugs that
selectively affect nicotinic and muscarinic receptors.
1. Muscarinic antagonists
The classical muscarinic
antagonists are atropine and the closely similar scopolamine. Atropine (Figure
9.12) will to some extent enter the brain and cause upheaval there as well –
this is reflected in the German name of the plant contain-ing it (Tollkirsche = "crazy-cherry").
It is used for local ap-plication to the eye (widening the pupil, relaxing the
ciliary muscle in diagnostic procedures), and during surgery to re-lax the
airways and suppress salivation, both of which will help to avoid respiratory
problems during narcosis. Iprat-ropium is used for oral and inhalation
treatment to relax the airways in asthma, and occasionally to speed up a slow
atri-oventricular node in the heart (cf. the preceding chapter). It is
preferred over atropin in most applications because it does not quite as easily
cross the blood brain barrier – by now, you should be able to tell why when
looking at the structure.
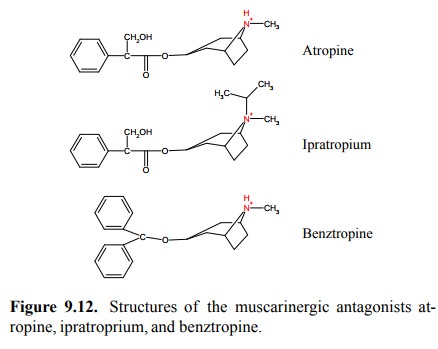
Benztropine is the most
hydrophobic one of the three drugs in Figure 9.12 and accordingly distributes
into the brain most easily. It is used as a component in the treatment of
Parkinson's disease; anticholinergic drugs augment12 the action of
dopaminergic drugs (such as L-DOPA) that re-main the mainstay of therapy in
this disease.
2. Nicotinic antagonists
Due to the molecular
differences between NAR at the neu-romuscular junction and the autonomic
ganglia, many nico-tinic antagonists are quite selective for one over the
other. Antagonists that are specific for the ganglia – `ganglion blockers' –
were among the first drugs to be used effec-tively for the treatment of
hypertonia. However, they have now been nearly entirely abandoned, which is due
to their rather numerous side effects which result from the block-ade of both
the sympathetic and the parasympathetic sys-tems. We have seen the structures
of hexamethonium and mecamylamine before in the chapter on pharmacokinetics –
you will note the similarity between their pharmacokinet-ic characteristics and
those described in this chapter for the other cholinergic agonists and
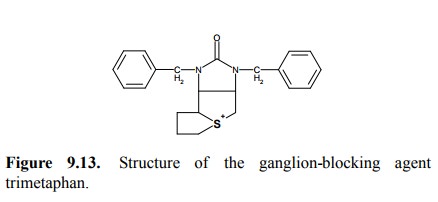
antagonists. Another inter-esting structure is that of trimethaphan (Figure
9.13), which is still sometimes used in a condition called hypertensive crisis.
In this molecule, the place of the quaternary amino group is taken by a
sulfonium ion, which consists of a sulfur atom bound to three aliphatic
substituents.
Nicotinergic antagonists with
specificity for the neuromus-cular junction will cause paralysis. These agents
are dis-cussed in the following section.
3. Muscle relaxants
Muscle relaxants suppress
muscle contraction in response to stimulation by the motoneurons. They may be
either antagonists or agonists of nicotinic acetylcholine receptors. Muscle
relaxants are routinely used in anesthesiology. To understand this application,
consider that narcosis serves two distinct purposes:
1. Suppression of pain perception,
2. Suppression of muscle motility – after all, we
don't want the patient to jump of the table and sleep walk around the operation
theater.
For the first purpose, it is sufficient to
abolish the activity of the higher centres in the brain, whereas the complete
abol-ishment of reflex motility requires suppression of more or less the entire
central nervous system. Although the latter can be achieved with narcotic gases
(such as diethylether and its more modern congeners) alone, it requires higher
dosages than are required for pain suppression only. The use of muscle
relaxants makes it possible to reduce the amount of narcotic gases needed, and
therefore to limit the associated risks and side effects.
4. Nicotinic antagonists used as muscle relaxants
The first muscle relaxant to
be discovered and clinically used was d-tubocurarine (Figure 9.14a), which is
found in curare, an arrow poison that was used by South American natives. Quite
a bit of imagination is required to spot any structural resemblance to
acetylcholine; d-tubocurarine is much larger and contains not one but two
quaternary amines within a rather large, rigid ring structure. The re-semblance
is more readily spotted with the more recent, synthetic compound pancuronium
(Figure 9.14a). This molecule has two acetylcholine moieties, embedded in a
larger structure that again looks rigid but otherwise does not have much
similarity to d-tubocurarine.
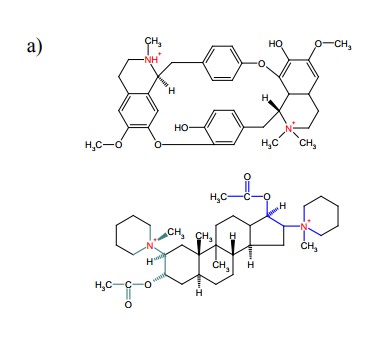
Both pancuronium and
d-tubocurarine act by binding to the NAR in a way competitive with
acetylcholine. In both cas-es, the presence of both positive charges is
important for ac-tivity. Yet, it is questionable whether both cationic groups
bind simultaneously to the two acetylcholine binding sites found (at the two α chains) of the NAR, since the distance between those should
substantially exceed the one between the two charges in the antagonist
molecules.
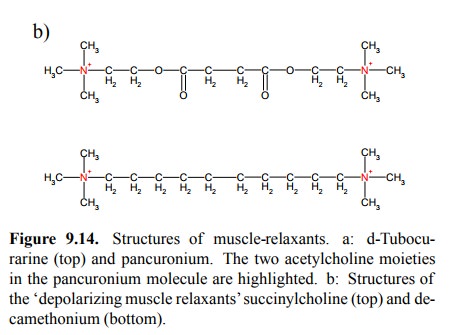
5. Depolarizing muscle relaxants
While both d-tubocurarine and
pancuronium prevent open-ing of the NAR, another class of muscle relaxants
effects neuromuscular blockade by triggering
the opening of NAR and, accordingly, membrane depolarization. Thus, these drugs
are not antagonists but agonists. In fact, this type of blockade – called
`depolarizing block' – can be achieved with acetylcholine itself, if used at
dosages exceeding those that occur physiologically13. The only agent
in widespread use is succinyl-bis-choline (also called succinylcholine);
this is just two
acetylcholine molecules joined end on (Fig-ure 9.14b). Another agonist is
decamethonium, which is a longer analog of hexamethonium; however, the latter
is an antagonist, not an agonist. One aspect of this blockade con-sists in the
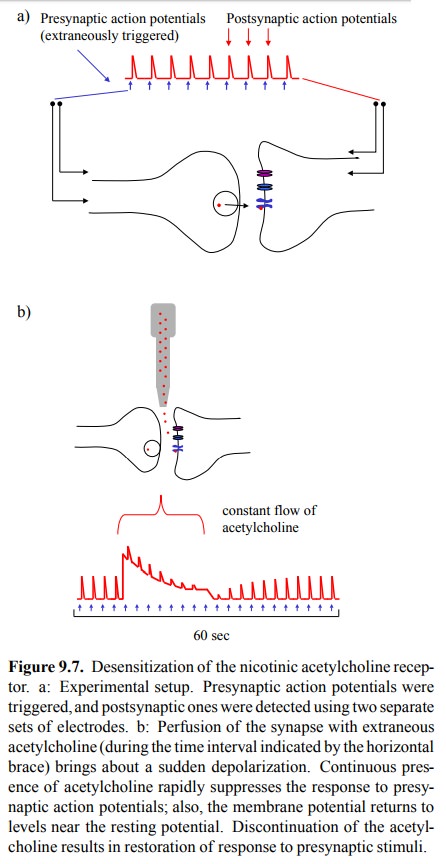
desensitization of the NAR – continuous pres-ence of acetylcholine or another
agonist will lead to accu-mulation of the receptor in the inactivated state
(cf. Figure 9.7, above). However, other mechanisms must be involved, too, which
follows from the following observations:
1. The membrane stays partially depolarized during
the presence of the agonist. If all receptors were fixed in the inactivated
state, the membrane should be completely repolarized.
2. If muscle cells have been cut off from their
neural in-put for a couple of weeks, they will express larger num-bers of NAR
molecules, even outside those membrane regions that were previously in contact
with the nerve cells. If acetylcholine or another depolarizing agent is applied
to these cells, they will respond with maximum contraction, and no blockade will
ensue. Thus, some regulatory mechanism that suppresses muscle contrac-tion upon
sustained membrane depolarization in nor-mal cells seems to be lost
concomitantly with the dener-vation.
Succinylcholine has a more
sustained action at the motor endplate than acetylcholine has because it is
insensitive to the acetylcholinesterase found in the synapse. There is,
however, a second variety of cholinesterase that cir-culates in the blood
plasma, also referred to as butyryl-cholinesterase14, which cleaves
succinylcholine within minutes. This moderately rapid inactivation makes it
pos-sible to control the degree of muscle relaxation by adjust-ing the infusion
rate of the agent; after discontinuation, the remaining succinylcholine will be
swiftly hydrolysed, and the block will subside.
Related Topics