Chapter: Biochemical Pharmacology : G protein-coupled receptors
Adrenergic receptor agonists and antagonists
Adrenergic receptor agonists
and antagonists
The first adrenergic receptor
types to be distinguished from each other were the adrenergic α- and β-receptors. Initially based on the different
cardiovascular effects of epinephrine and norepinephrine, this distinction was
borne out more clearly with the synthetic β-selective agent isoproterenol. Furthermore,
subtypes of both α- and β-receptors can be distinguished by selective
agonists (Figure 10.5).
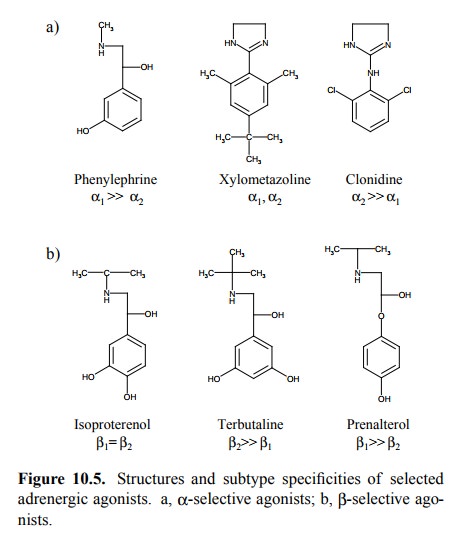
1. Physiological effects of α- and β-selective adrenergic
agonists
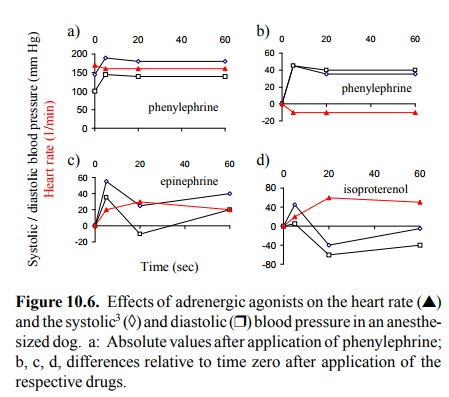
The different cardiovascular
effects of α- and β-receptors are illustrated in the experiment in
Figure 10.6. Phenyle-phrine, an α-selective agent, causes vasoconstriction and
accordingly a rise in blood pressure. The heart has few α-receptors, so phenylephrine does not accelerate the heart rate. Rather, the heart rate drops a little, due to an auto-nomic
physiological reflex triggered by the increased blood pressure2.
Conversely, the β-selective agonist isoproterenol speeds up the
heart rate quite significantly, yet neverthe-less causes the blood pressure to
drop, which is due to va-sodilatation. The initial brief spike of the blood pressure
is probably caused by delayed distribution of the drug into the skeletal
muscle, where most of the vascular β2-receptors are located. Vasodilatation will therefore lag behind
the stimulatory effect on the heart, since the heart is perfused much more
strongly than the skeletal muscle, especially at rest. Epinephrine, which
stimulates both α- and β-recep-tors, shows an intermediate response.
2. Physiological effects of α2-adrenergic agonists
The vasopressor response seen
with phenylephrine is par-ticularly strong because it is an α1-selective
agonist. α2-Ag-onists
actually lower the blood pressure, which is in ac-cord with the role of the α2-receptors
in presynaptic inhi-bition. This inhibition is shown in Figure 10.7a: The re
lease of radioactively labeled norepinephrine from the nerve terminals is
reduced by about 80% with an α2 agonist (UK 14,304). In the presence of α-receptor blockers (rau-wolscine or phentolamine), we see a right
shift of the dose response curve, which confirms the receptor-specific mode of
action of this effect.
Further confirmation of the
role of the α2-receptor
comes from gene knockout experiments (Figure 10.7b). There are actually three
discernible subtypes (or subsubtypes – have as many subs as you please) of the α2
receptor, named A/D, B and C, with distinct genes on different chromosomes. In
the particular cell type used in the experiment shown, the A/D subtype was
evidently responsible for most of the inhibition of transmitter release.
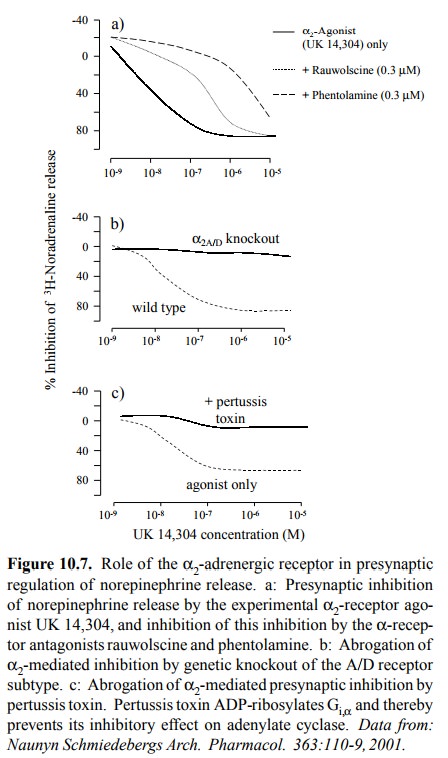
While this inhibition is very nice and unequivocal,
the in-tracellular events that occur subsequent to α2
receptor stim-ulation are less clearly defined. The experiment depicted in
Figure 10.7c suggests that indeed, as stated earlier, the α2-receptor
acts by lowering cAMP. Pertussis toxin– the major toxin secreted by the bacterium
Bordetella pertussis, the causative agent of whooping cough – is able to cross
the cell membrane and then covalently modify4 the α-subunit ot the Gi protein that normally inhibits
adenylate cyclase when activated by the α2 receptor.
Although pertussis toxin is a
popular tool in signal transduc-tion research, it acts on several G proteins
other than Gi as well, so that the implication of cAMP by toxin
inhibition is not cogent. In fact, experimental evidence is also available that
throws the role of cAMP in α2-mediated effects into question. You will recall that the release
of insulin from pancreatic islet cells is closely similar to neurotransmitter
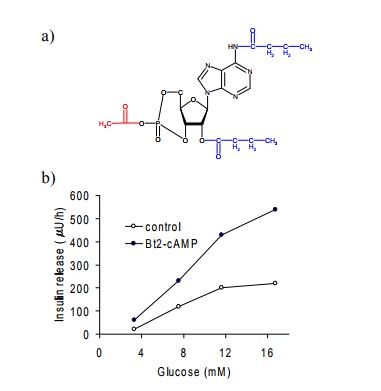
exocytosis. It is stimulated by glucose and further ampli-fied by cAMP. The
effect of cAMP can be studied using the cell-permeant synthetic analog
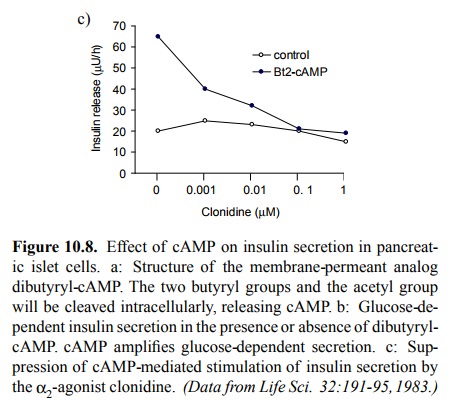
dibutyryl-cAMP (Figure 10.8a). Intracellularly, the butyryl and acetyl groups
of this `pro-drug' will get cleaved off to release cAMP itself, and in Figure
10.8b you can see that the cAMP released indeed increases insulin secretion.
However, at sufficiently high concentrations, clonidine suppresses the
increased insulin release caused by cAMP (Figure 10.8c). This suggests that the
effect of the α2-receptor
stimulation must be located somewhere downstream of adenylate cyclase and cAMP.
α2-Receptor
stimulation has measurable effects on Ca++ and K+ channels, which are involved in insulin
secretion.
How these effects relate to cAMP or to the βγ-subunits of Gi is not known with certainty.
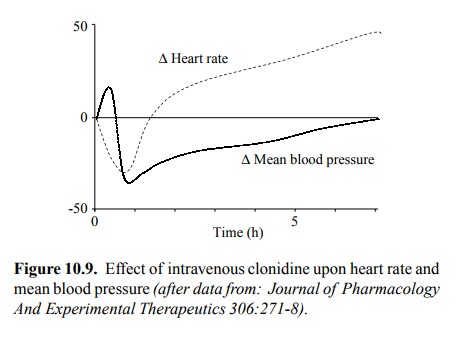
Figure 10.9 illustrates the
blood-pressure lowering effect of clonidine. As with isoproterenol (cf. Figure
10.6), the ear-ly effects go into the opposite direction, but here this phase
lasts minutes rather than seconds, which probably reflects the need of the drug
to cross the blood brain barrier to elicit the drop in blood pressure. In this
case, we see a very strong reflectory increase of the heart rate that finally
negates the drop in blood pressure. This type of counter-regulation is very
common. Effective treatment of hypertension there-fore frequently requires the
combined use of several drugs with complementary modes of action. For example,
the re-flectory increase in heart rate could effectively by coun-tered by the
simultaneous application of β-receptor antago-nists.
3. β-Adrenergic agonists
β1-selective
agonists such as prenalterol (Figure 10.5) or dobutamine are used to stimulate heart
contractility and heart rate in patients that exhibit too low blood pressure. β2-Selective
agents such as terbutaline find application for relaxation and dilatation of the bronchi in
the treat-ment of asthma or chronic obstructive lung disease, and in the
suppression of premature labour. The mechanism of β-receptor-induced relaxation of smooth muscle – cAMP-mediated
phosphorylation and inhibition of myosin light chain kinase – has been
described before.
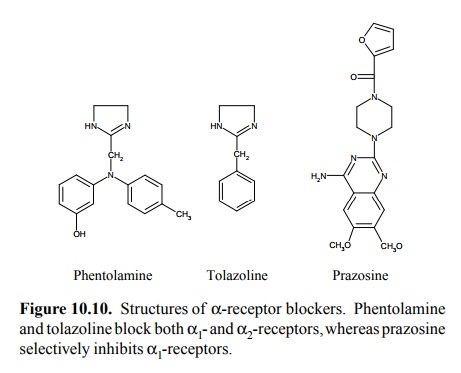
4. α-Adrenergic antagonists
Like agonists, adrenergic
receptor antagonists come in var-ious degrees of type and subtype selectivity.
Among the α-receptor antagonists, phentolamine and tolazoline (Fig-ure 10.10)
act on both α1- and α2-receptors.
α2-Selective
antagonists do exist (e.g. yohimbine) but don't have ma-jor therapeutical
applications. Considering the physiolog-ical role of α2
receptors, it is clear that α1-selective antag-onists such as prazosine are preferable for use in
the thera-py of hypertension. In addition to reversible blockers, irre-versible
(covalent) blockers are available for particular sit-uations such as
adrenaline-producing tumours (cf. the dis-cussion of phenoxybenzamine in the
chapter on pharmaco-dynamics).
5. β-Adrenergic antagonists
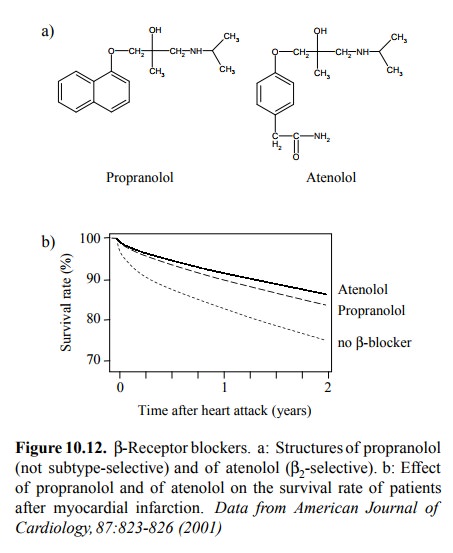
The practically most
important class of drugs directly act-ing on any adrenergic receptor are the β-blockers (Figure 10.12). They are mainly used to reduce the
workload on a heart subject to impaired perfusion due to atherosclerotic blood
vessel obliteration. The most severe consequence of this impaired perfusion is
myocardial infarction, which consists in the irreversible damage to the regions
of heart muscle tissue downstream of an obliterated artery. Patients
experiencing the first such event are at considerable con-tinuous risk of
repeated attacks. Accordingly, the number of survivors in this group of
patients decreases substantial-ly with time. β-Blockers are among the very few agents that
afford them a substantial and indubitable benefit, in terms of life expectancy.
While in Figure 10.12b atenolol appears somewhat superior to propanolol, the
effect of an-other β2-selective blocker (metoprolol) was virtually indis-tinguishable
from that of propranolol (not shown). There is thus far no conclusive evidence
that β2-selective
antagonists really are more effective than those that are not subtype-specific.
Related Topics