Chapter: Basic & Clinical Pharmacology : Antiseizure Drugs
Phenytoin
PHENYTOIN
Phenytoin
is the oldest nonsedative antiseizure drug, introduced in 1938 after a
systematic evaluation of compounds such as phe-nobarbital that altered
electrically induced seizures in laboratory animals. It was known for decades
as diphenylhydantoin.
Chemistry
Phenytoin
is a diphenyl-substituted hydantoin with the structure shown. It has much lower
sedative properties than compounds with alkyl substituents at the 5 position. A
more soluble prodrug.of phenytoin, fosphenytoin,
is available for parenteral use; this phosphate ester compound is rapidly
converted to phenytoin in the plasma.
Mechanism of Action
Phenytoin
has major effects on several physiologic systems. It alters Na+, K+,
and Ca2+ conductance, membrane potentials, and the concentrations of
amino acids and the neurotransmitters nor-epinephrine, acetylcholine, and γ-aminobutyric acid
(GABA). Studies with neurons in cell culture show that phenytoin blocks
sustained high-frequency repetitive firing of action potentials (Figure 24–4).
This effect is seen at therapeutically relevant con-centrations. It is a
use-dependent effect on Na+
conductance, arising from preferential binding to—and prolonga-tion of—the
inactivated state of the Na+ channel. This effect is also seen with
therapeutically relevant concentrations of carbam-azepine, lamotrigine, and
valproate and probably contributes to their antiseizure action in the
electroshock model and in partial seizures. Phenytoin also blocks the
persistent Na+ current, as do several other AEDs including
valproate, topiramate, and ethosux-imide.
In
addition, phenytoin paradoxically causes excitation in some cerebral neurons. A
reduction of calcium permeability, with inhi-bition of calcium influx across
the cell membrane, may explain the ability of phenytoin to inhibit a variety of
calcium-induced secre-tory processes, including release of hormones and
neurotransmit-ters. Recording of excitatory and inhibitory postsynaptic
potentials show that phenytoin decreases the synaptic release of glutamate.

and
enhances the release of GABA. The mechanism of phenytoin’s action probably
involves a combination of actions at several levels. At therapeutic
concentrations, the major action of phenytoin is to block Na+
channels and inhibit the generation of rapidly repetitive action potentials.
Presynaptic actions on glutamate and GABA release probably arise from actions
other than those on voltage-gated Na+ channels.
Clinical Uses
Phenytoin
is effective against partial seizures and generalized tonic-clonic seizures. In
the latter, it appears to be effective against attacks that are either primary
or secondary to another seizure type.
Pharmacokinetics
Absorption
of phenytoin is highly dependent on the formulation of the dosage form.
Particle size and pharmaceutical additives affect both the rate and the extent
of absorption. Absorption of phenytoin sodium from the gastrointestinal tract
is nearly com-plete in most patients, although the time to peak may range from
3 to 12 hours. Absorption after intramuscular injection is unpre-dictable, and
some drug precipitation in the muscle occurs; this route of administration is
not recommended for phenytoin. In contrast, fosphenytoin, a more soluble
phosphate prodrug of phe-nytoin, is well absorbed after intramuscular
administration.
Phenytoin
is highly bound to plasma proteins. The total plasma level decreases when the
percentage that is bound decreases, as in uremia or hypoalbuminemia, but
correlation of free levels with clinical states remains uncertain. Drug
concentration in cere-brospinal fluid is proportionate to the free plasma
level. Phenytoin accumulates in brain, liver, muscle, and fat.
Phenytoin
is metabolized to inactive metabolites that are excreted in the urine. Only a
very small proportion of the dose is excreted unchanged.
The
elimination of phenytoin is dose-dependent. At very low blood levels, phenytoin
metabolism follows first-order kinetics. However, as blood levels rise within
the therapeutic range, the maximum capacity of the liver to metabolize
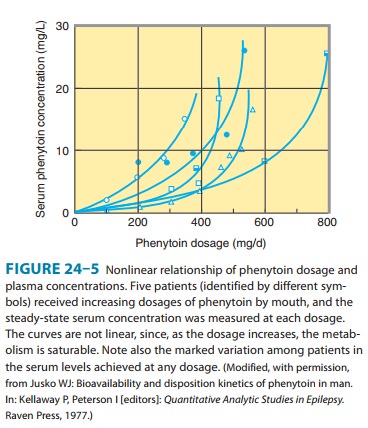
phenytoin is approached. Further increases in dosage, though relatively small,
may produce very large changes in phenytoin concentrations (Figure 24–5). In
such cases, the half-life of the drug increases markedly, steady state is not
achieved in routine fashion (since the plasma level continues to rise), and
patients quickly develop symp-toms of toxicity.
The
half-life of phenytoin varies from 12 to 36 hours, with an average of 24 hours
for most patients in the low to mid therapeu-tic range. Much longer half-lives
are observed at higher concentra-tions. At low blood levels, it takes 5–7 days
to reach steady-state blood levels after every dosage change; at higher levels,
it may be 4–6 weeks before blood levels are stable.
Therapeutic Levels & Dosage
The
therapeutic plasma level of phenytoin for most patients is between 10 and 20
mcg/mL. A loading dose can be given either orally or intravenously; the latter,
using fosphenytoin, is the method of choice for convulsive status epilepticus
(discussed later). When oral therapy is started, it is common to begin adults
at a dosage of 300 mg/d, regardless of body weight. This may be acceptable in
some patients, but it frequently yields steady-state blood levels below 10
mcg/mL, which is the mini-mum therapeutic level for most patients. If seizures
continue, higher doses are usually necessary to achieve plasma levels in the
upper therapeutic range. Because of its dose-dependent kinet-ics, some toxicity
may occur with only small increments in dos-age. The phenytoin dosage should be
increased each time by only 25–30 mg in adults, and ample time should be
allowed for the new steady state to be achieved before further increasing the
dosage. A common clinical error is to increase the dosage directly from 300
mg/d to 400 mg/d; toxicity frequently occurs at a variable time thereafter. In
children, a dosage of 5 mg/kg/d should be followed by readjustment after
steady-state plasma levels are obtained.
Two types of oral phenytoin sodium are currently available in the USA, differing in their respective rates of dissolution; one is absorbed rapidly and one more slowly. Only the slow-release extended-action formulation can be given in a single daily dos-age, and care must be used when changing brands (see Preparations Available). Although a few patients being given phenytoin on a long-term basis have been proved to have low blood levels from poor absorption or rapid metabolism, the most common cause of low levels is poor compliance. Fosphenytoin sodium is available for intravenous or intramuscular use and replaces intravenous phenytoin sodium, a much less soluble form of the drug.
Drug Interactions & Interference with Laboratory Tests
Drug
interactions involving phenytoin are primarily related to protein binding or to
metabolism. Since phenytoin is 90% bound to plasma proteins, other highly bound
drugs, such as phenylbuta-zone and sulfonamides, can displace phenytoin from
its binding site. In theory, such displacement may cause a transient increase
in free drug. A decrease in protein binding—eg, from hypoalbumin-emia—results
in a decrease in the total plasma concentration of drug but not the free
concentration. Intoxication may occur if efforts are made to maintain total
drug levels in the therapeutic range by increasing the dose. The protein
binding of phenytoin is decreased in the presence of renal disease. The drug
has an affinity for thyroid-binding globulin, which confuses some tests of
thyroid function; the most reliable screening test of thyroid function in
patients taking phenytoin appears to be measurement of thyroid-stimulating
hormone (TSH).
Phenytoin
has been shown to induce microsomal enzymes responsible for the metabolism of a
number of drugs. Autostimu-lation of its own metabolism, however, appears to be
insignificant.
Toxicity
Dose-related
adverse effects caused by phenytoin are often similar to those caused by other
antiseizure drugs in this group, making differentiation difficult in patients
receiving multiple drugs. Nystagmus occurs early, as does loss of smooth
extraocular pursuit movements, but neither is an indication for decreasing the
dose. Diplopia and ataxia are the most common dose-related adverse effects requiring
dosage adjustment; sedation usually occurs only at considerably higher levels.
Gingival hyperplasia and hirsutism occur to some degree in most patients; the
latter can be especially unpleasant in women. Long-term use is associated in
some patients with coarsening of facial features and with mild periph-eral
neuropathy, usually manifested by diminished deep tendon reflexes in the lower
extremities. Long-term use may also result in abnormalities of vitamin D
metabolism, leading to osteomalacia. Low folate levels and megaloblastic anemia
have been reported, but the clinical importance of these observations is
unknown.
Idiosyncratic
reactions to phenytoin are relatively rare. A skin rash may indicate
hypersensitivity of the patient to the drug. Fever may also occur, and in rare
cases the skin lesions may be severe and exfoliative. Lymphadenopathy may be
difficult to distinguish from malignant lymphoma, and although some studies
suggest a causal relationship between phenytoin and Hodgkin’s disease, the data
are far from conclusive. Hematologic complications are exceedingly rare,
although agranulocytosis has been reported in combination with fever and rash.
Related Topics