Chapter: Biochemistry: The Three-Dimensional Structure of Proteins
Prions and Disease
Prions and Disease
It has
been established that the causative agent of mad-cow disease (also known as
bovine spongiform encephalopathy or BSE), as well as the related diseases
scrapie in sheep, chronic wasting disease (CWD) in deer and elk, and human
spongiform encephalopathy (kuru and Creutzfeldt-Jakob disease) in humans, is a
small (28-kDa) protein called a prion. (Note that biochemists tend to call the
unit of atomic mass the dalton,
abbreviated Da.) Prions are natural glycoproteins found in the cell membranes
of nerve tissue. Recently the prion protein has been found in the cell membrane
of hematopoietic stem cells, precursors to the cells of the bloodstream, and
there is some evidence that the prion helps guide cell maturation. The disease
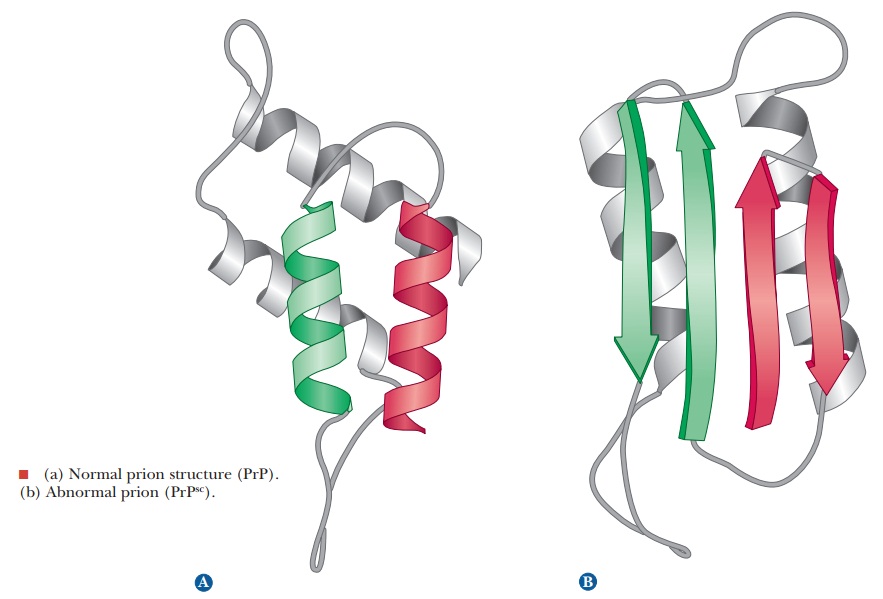
state comes about when the normal form of the prion protein, PrP (Figure a),
folds into an incorrect form called PrPsc (Figure
b). The abnormal form of the prion protein is able to convert other, normal
forms into abnormal forms. As was recently discovered, this change can be
propagated in nervous tissue. Scrapie had been known for years, but it had not
been known to cross species barriers. Then an outbreak of mad-cow disease was
shown to have followed the inclusion of sheep remains in cattle feed. It is now
known that eating tainted beef from animals with mad-cow disease can cause
spongiform encephalopathy, now known as new variant Creutzfeldt-Jakob disease
(vCJD), in humans. The normal prions have a large percentage of α-helix, but
the abnormal forms have more β-pleated sheets. Notice that in this case the
same protein (a single, well-defined sequence) can exist in alternative forms.
These β-pleated sheets in the abnormal proteins interact between protein
molecules and form insoluble plaques, a fate also seen in Alzheimer’s disease.
Ingested abnormal prions use macrophages from the immune system to travel in
the body until they come in contact with nerve tissue. They can then propagate
up the nerves until they reach the brain.
This
mechanism was a subject of considerable controversy when it was first proposed.
A number of scientists expected that a slow-acting virus would be found to be
the ultimate cause of these neurological diseases. A susceptibility to these
diseases can be inherited, so some involvement of DNA (or RNA) was also
expected. Some went so far as to talk about “heresy” when Stanley Prusiner
received the 1997 Nobel Prize in medicine for his discovery of prions, but
substantial evidence shows that prions are themselves the infectious agent and
that no virus or bacteria are involved. It now appears that genes for
susceptibility to the incorrect form exist in all vertebrates, giving rise to
the observed pattern of disease transmission, but many individuals with the
genetic susceptibility never develop the disease if they do not come in contact
with abnormal prions from another source.

Further
studies have shown that all of the humans who showed symptoms of vCJD had the
same amino acid substitution in their prions, a substitution of a methionine at
position 129, now known to confer extreme sensitivity to the disease. While the
cases of vCJD are decreasing, some scientists warn that the lag between the
increase in mad-cow disease in Europe and the peak in human infections was too
short. They feel that the peak that is now declining represents the population
of humans that had the most severe mutation leading to the extreme sensitivity.
It is possible that other populations of humans have different muta-tions and
lesser sensitivities. These other populations could have longer incubation
times, so the vCJD threat may not be over yet.
Related Topics