Chapter: Biochemistry: The Three-Dimensional Structure of Proteins
Forces Involved in Tertiary Structures
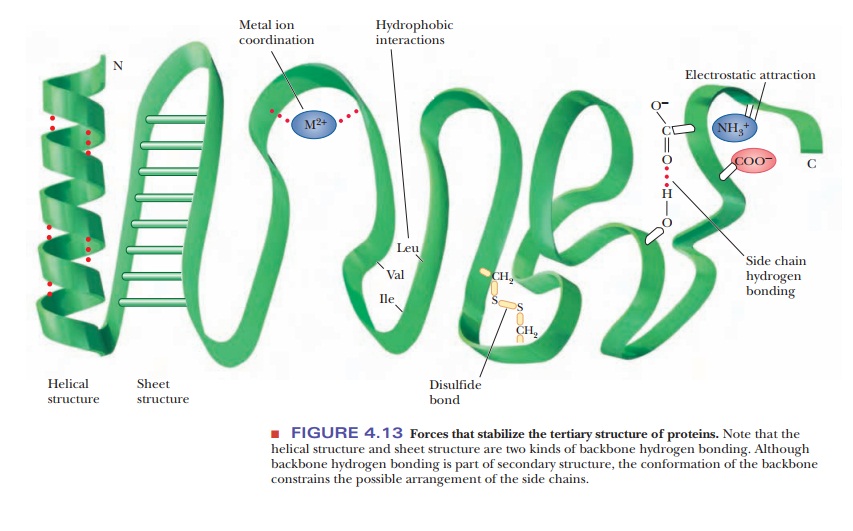
Forces Involved in Tertiary Structures
Many types of forces and interactions play a role in holding a protein together in its correct, native conformation. Some of these forces are covalent, but many are not. The primary structure of a protein-the order of amino acids in the polypeptide chain-depends on the formation of peptide bonds, which are covalent. Higher-order levels of structure, such as the conformation of the backbone (secondary structure) and the positions of all the atoms in the protein (tertiary structure), depend on noncovalent interactions. If the protein consists of several subunits, the interaction of the subunits also depends on noncovalent interactions. Noncovalent stabilizing forces contribute to the most stable structure for a given protein, the one with the lowest energy.
Several types of hydrogen bonding occur in proteins. Backbone hydrogen bonding is a major determinant of secondary structure; hydrogen bonds between the side chains of amino acids are also possible in proteins. Nonpolar resi-dues tend to cluster together in the interior of protein molecules as a result of hydrophobic interactions. Electrostatic attraction between oppositely charged groups, which frequently occurs on the surface of the molecule, results in such groups being close to one another. Several side chains can be complexedto a single metal ion. (Metal ions also occur in some prosthetic groups.)
In addition to these noncovalent interactions, disulfide bonds form covalent links between the side chains of cysteines. When such bonds form, they restrict the folding patterns available to polypeptide chains. There are specialized labo-ratory methods for determining the number and positions of disulfide links in a given protein. Information about the locations of disulfide links can then be combined with knowledge of the primary structure to give the complete covalentstructure of the protein. Note the subtle difference here: The primary structureis the order of amino acids, whereas the complete covalent structure also speci-fies the positions of the disulfide bonds (Figure 4.13).
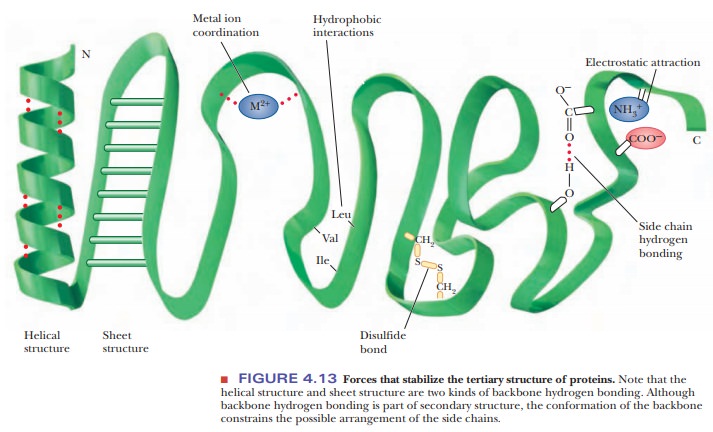
Not every protein necessarily exhibits all possible structural features of the kinds just described. For instance, there are no disulfide bridges in myoglobin and hemoglobin, which are oxygen-storage and transport proteins and classic examples of protein structure, but they both contain Fe(II) ions as part of a prosthetic group. In contrast, the enzymes trypsin and chymotrypsin do not contain complexed metal ions, but they do have disulfide bridges. Hydrogen bonds, electrostatic interactions, and hydrophobic interactions occur in most proteins.
The three-dimensional conformation of a protein is the result of the inter-play of all the stabilizing forces. It is known, for example, that proline does not fit into an α-helix and that its presence can cause a polypeptide chain to turn a corner, ending an α-helical segment. The presence of proline is not, however, a requirement for a turn in a polypeptide chain. Other residues are rou-tinely encountered at bends in polypeptide chains. The segments of proteins at bends in the polypeptide chain and in other portions of the protein that are not involved in helical or pleated-sheet structures are frequently referred to as “random” or “random coil.” In reality, the forces that stabilize each protein are responsible for its conformation.
How can the three-dimensional structure of a protein be determined?
The experimental technique used to determine the tertiary structure of a protein is X-ray crystallography. Perfect crystals of some proteins can be grown under carefully controlled conditions. In such a crystal, all the individual protein molecules have the same three-dimensional conformation and the same orientation. Crystals of this quality can be formed only from proteins of very high purity, and it is not possible to obtain a structure if the protein cannot be crystallized.
When a suitably pure crystal is exposed to a beam of X rays, a diffraction pat-tern is produced on a photographic plate (Figure 4.14a) or a radiation counter.The pattern is produced when the electrons in each atom in the molecule scat-ter the X rays. The number of electrons in the atom determines the intensity of its scattering of X rays; heavier atoms scatter more effectively than lighter atoms. The scattered X rays from the individual atoms can reinforce each other or cancel each other (set up constructive or destructive interference), giving rise to the characteristic pattern for each type of molecule. A series of diffrac-tion patterns taken from several angles contains the information needed to determine the tertiary structure. The information is extracted from the diffrac-tion patterns through a mathematical analysis known as a Fourier series. Many thousands of such calculations are required to determine the structure of a pro-tein, and even though they are performed by computer, the process is a fairly long one. Improving the calculation procedure is a subject of active research.
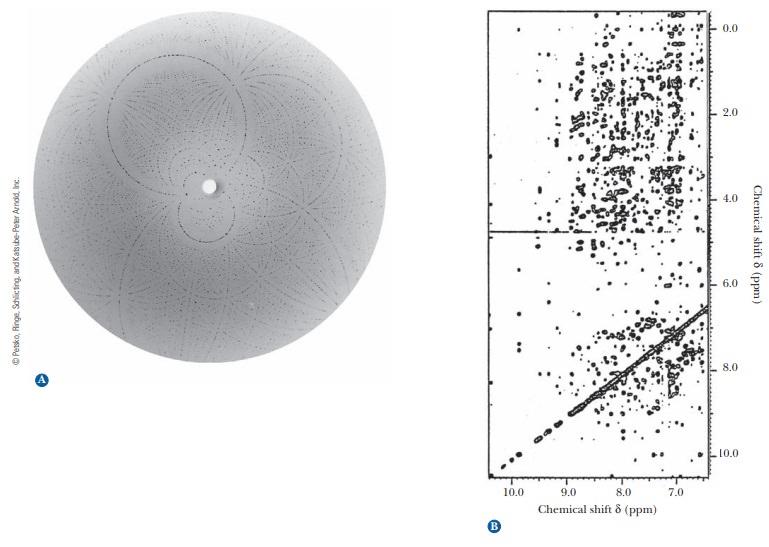
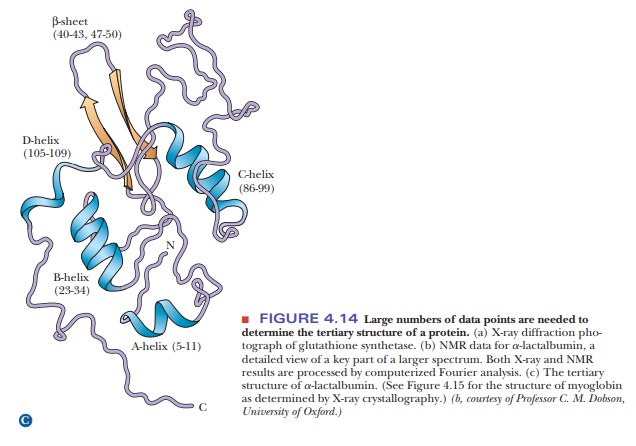
Another technique that supplements the results of X-ray diffraction has come into wide use in recent years. It is a form of nuclear magnetic resonance(NMR) spectroscopy. In this particular application of NMR, called2-D(twodimensional) NMR, large collections of data points are subjected to computer analysis (Figure 4.14b). Like X-ray diffraction, this method uses a Fourier series to analyze results. It is similar to X-ray diffraction in other ways: It is a long pro-cess, and it requires considerable amounts of computing power and milligram quantities of protein. One way in which 2-D NMR differs from X-ray diffraction is that it uses protein samples in aqueous solution rather than crystals. This environment is closer to that of proteins in cells, and thus it is one of the main advantages of the method. The NMR method most widely used in the deter-mination of protein structure ultimately depends on the distances between hydrogen atoms, giving results independent of those obtained by X-ray crystal-lography. The NMR method is undergoing constant improvement and is being applied to larger proteins as these improvements progress.
Related Topics