Chapter: Biochemistry: The Three-Dimensional Structure of Proteins
Secondary Structure of Proteins
![]()
![]() Secondary Structure of Proteins
Secondary Structure of Proteins
The
secondary structure of proteins is the hydrogen-bonded arrangement of the
backbone of the protein, the polypeptide chain. The nature of the bonds in the
peptide backbone plays an important role here. Within each amino acid residue
are two bonds with reasonably free rotation: (1) the bond between the α-carbon
and the amino nitrogen of that residue and (2) the bond between the α-carbon
and the carboxyl carbon of that residue. The combination of the planar peptide
group and the two freely rotating bonds has important implications for the
three-dimensional conformations of peptides and proteins. A peptide-chain
backbone can be visualized as a series of playing cards, each card representing
a planar peptide group. The cards are linked at opposite corners by swivels,
representing the bonds about which there is considerable freedom

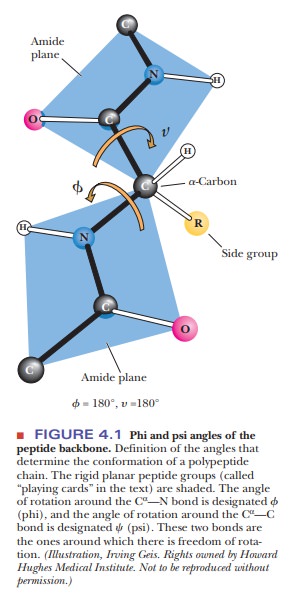
The side chains also play a vital role in determining
the three-dimensional shape of a protein, but only the backbone is considered
in the secondary structure. The angles Φ (phi)
and Ψ (psi), frequently called Ramachandran angles (after their
originator, G. N. Ramachandran), are used to designate rotations around the C-N
and C-C bonds, respectively. The conformation of a protein backbone can be
described by specifying the values of Φ and Ψ for
each residue (–180° to 180°). Two kinds of secondary structures that occur
frequently in proteins are the repeating α-helix and β-pleated sheet (or β-sheet)
hydrogen-bonded structures. The Φ and Ψ angles
repeat themselvesin contiguous amino acids in regular secondary structures. The
α-helix and β-pleated sheet are not the
only possible secondary structures, but they are by far the most important and
deserve a closer look.
Periodic Structures in Protein Backbones
The α-helix
and β-pleated sheet are periodic structures; their features repeat at
regular intervals. The α-helix is rodlike and
involves only one polypeptide chain. The β-pleated
sheet structure can give a two-dimensional array and can involve one or more
polypeptide chains.
Why is the A-helix so prevalent?
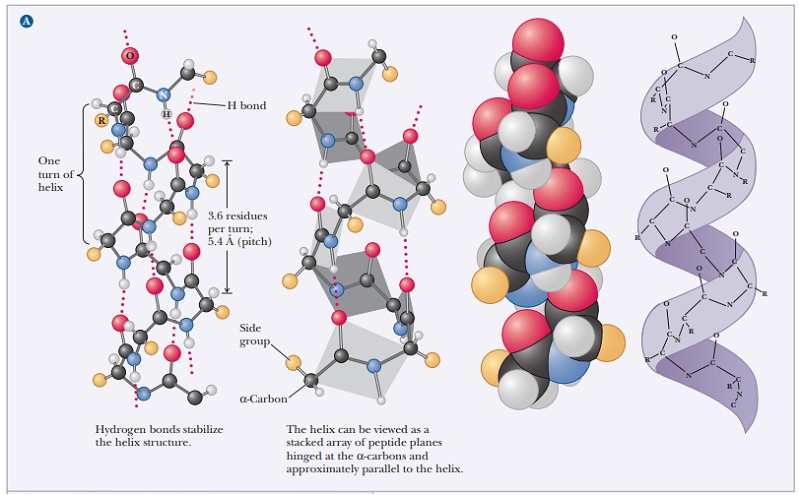
The α-helix
is stabilized by hydrogen bonds parallel to the helix axis within the backbone
of a single polypeptide chain. Counting from the N-terminal end, the C-O group
of each amino acid residue is hydrogen bonded to the N-H group of the amino acid four residues away from
it in the covalently bonded sequence. The helical conformation allows a linear
arrangement of the atoms involved in the hydrogen bonds, which gives the bonds
maximum strength and thus makes the helical conformation very stable. There are
3.6 residues for each turn of the helix, and the pitch of the helix (the linear distance between corresponding
points on successive turns) is 5.4 Å (Figure 4.2).
The angstrom unit, 1 Å 5 10–8 cm 5 10–10
m, is convenient for inter-atomic distances in molecules, but it is not a
Système International (SI) unit. Nanometers (1 nm 5 10–9 m) and picometers (1 pm 5 10–12 m) are the SI units used for interatomic
distances. In SI units, the pitch of the α-helix is 0.54 nm or 540 pm. Figure
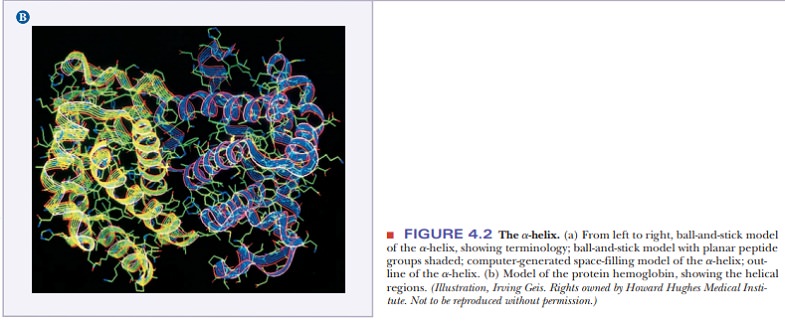
4.3 shows the structures of two proteins with a high degree of α-helical
content.
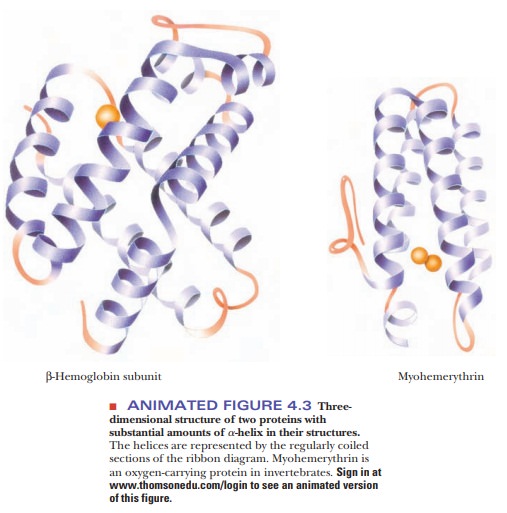
Proteins have varying amounts of α-helical
structures, varying from a few percent to nearly 100%. Several factors can
disrupt the α-helix. The amino acid proline creates a bend in the backbone
because of its cyclic structure. It
cannot fit into the α-helix because (1) rotation around the bond between the
nitrogen and the α-carbon is severely restricted, and (2) proline’s α-amino
group cannot participate in intrachain hydrogen bonding. Other localized
factors involving the side chains include strong electrostatic repulsion owing
to the proximity of several charged groups of the same sign, such as groups of
positively charged lysine and arginine residues or groups of negatively charged
glutamate and aspartate residues. Another possibility is crowding (steric
repulsion) caused by the proximity of several bulky side chains. In the α-helical
conformation, all the side chains lie outside the helix; there is not enough
room for them in the interior. The α-carbon is just outside the helix, and crowding
can occur if it is bonded to two atoms other than hydrogen, as is the case with
valine, isoleucine, and threonine.
How is the B-sheet different from the A-helix?
The arrangement of atoms in the β-pleated sheet conformation differs markedly from that in the α-helix. The peptide backbone in the β-sheet is almost completely extended. Hydrogen bonds can be formed between different parts of a single chain that is doubled back on itself (intrachain bonds) or between different chains (interchain bonds). If the peptide chains run in the same direction (i.e., if they are all aligned in terms of their N-terminal and C-terminal ends), a parallel pleated sheet is formed.
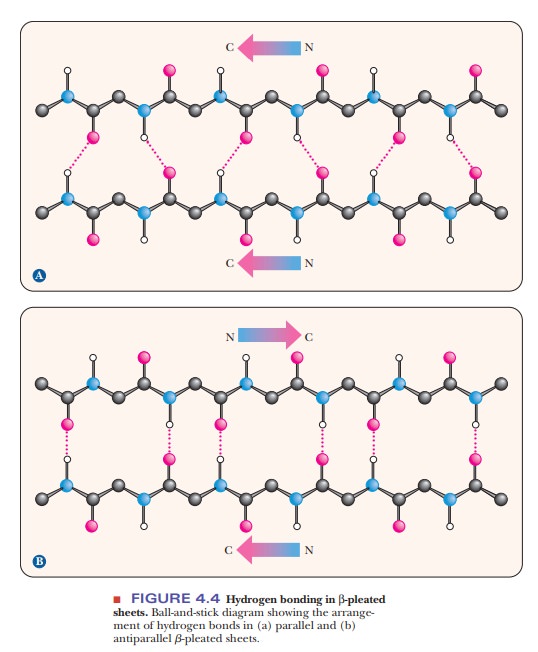
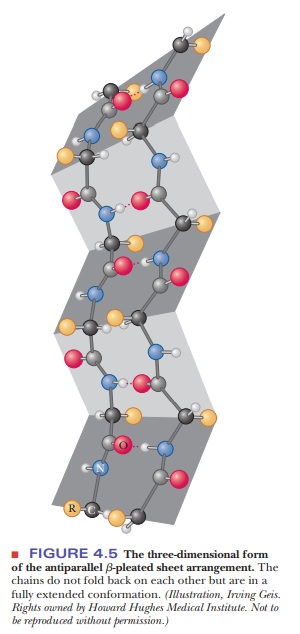
When alternating chains run in opposite directions, an antiparallel pleated sheet is formed (Figure 4.4). The hydrogen bonding between peptide chains in the β-pleated sheet gives rise to a repeated zigzag structure; hence, the name “pleated sheet” (Figure 4.5). Note that the hydrogen bonds are perpendicular to the direction of the protein chain, not parallel to it as in the α-helix.
![]()
![]()
Irregularities in Regular Structures
Other helical structures are found in proteins.
These are often found in shorter stretches than with the α-helix, and they
sometimes break up the regular nature of the α-helix. The most common is the 310
helix, which has three residues per turn and 10 atoms in the ring formed by
making the hydrogen bond. Other common helices are designated 27 and
4.416, following the same nomenclature as the 310 helix.
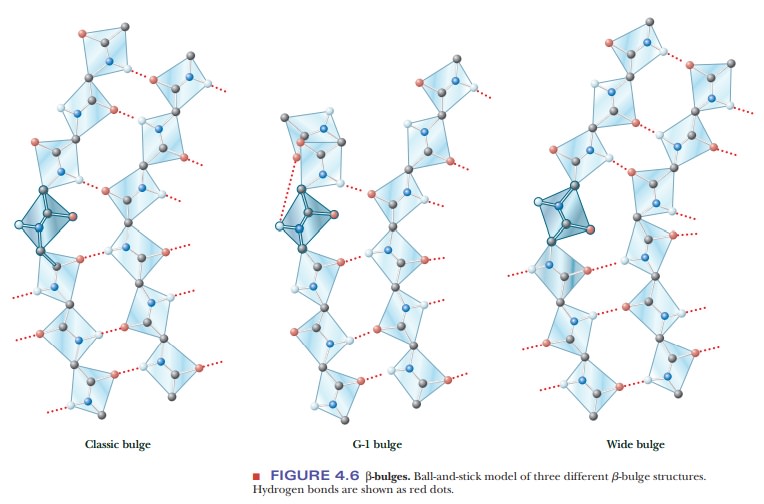
A β-bulge is a common nonrepetitive
irregularity found in antiparallel β-sheets. It occurs between two normal β-structure
hydrogen bonds and involves two residues on one strand and one on the other.
Figure 4.6 shows typical β-bulges.
Protein folding requires that the peptide
backbones and the secondary structures be able to change directions. Often a
reverse turn marks a transition between one secondary structure and another.
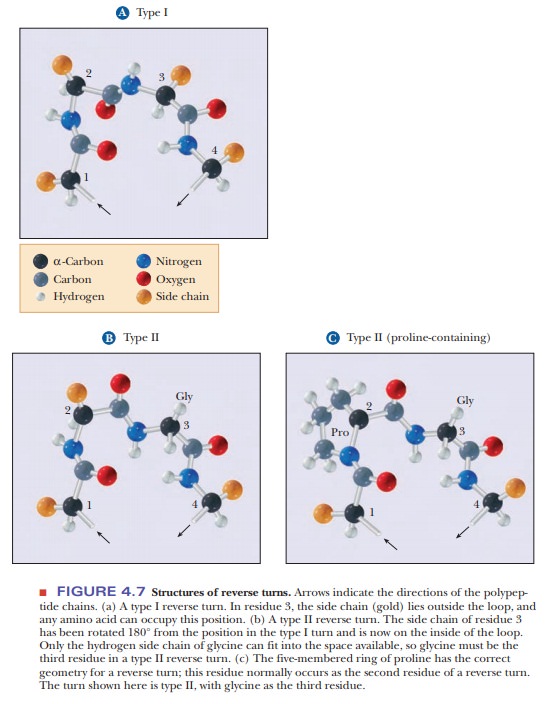
For steric (spatial) reasons, gly-cine is frequently encountered in reverse turns, at which the polypeptide
chain changes direction; the single hydrogen of the side chain prevents
crowding (Figures 4.7a and 4.7b). Because the cyclic structure of proline has
the correct geometry for a reverse turn, this amino acid is also frequently
encountered in such turns (Figure 4.7c).
Supersecondary Structures and Domains
The α-helix,
β-pleated sheet, and other secondary structures are combined in many ways as
the polypeptide chain folds back on itself in a protein. The combination of α-
and β-strands produces various kinds of supersecondary structures in proteins.
The most common feature of this sort is the bab unit, in which two parallel strands of β-sheet are connected by a
stretch of α-helix (Figure 4.8a). An aa unit
(helix-turn-helix) consists of two antiparallel α-helices (Figure 4.8b). In
such an arrangement, energetically favorable contacts exist between the side
chains in the two stretches of helix. In a β-meander, an antiparallel sheet is formed by a series of tight
reverse turns connecting stretches of the polypeptide chain (Figure 4.8c).
Another kind of antiparallel sheet is formed when the polypeptide chain doubles
back on itself in a pattern known as the Greek
key, named for a decorative design found on pottery from the
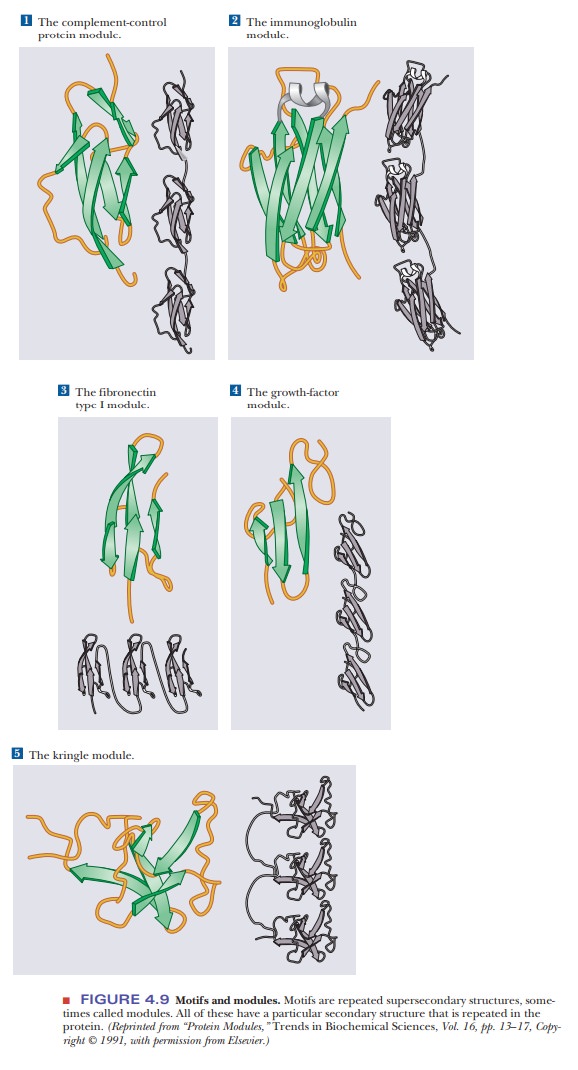
classicalperiod (Figure 4.8e). A motif
is a repetitive supersecondary structure. Some of the common smaller motifs are
shown in Figure 4.9. These smaller motifs can often be repeated and organized
into larger motifs. Protein sequences that allow for a β-meander or Greek key
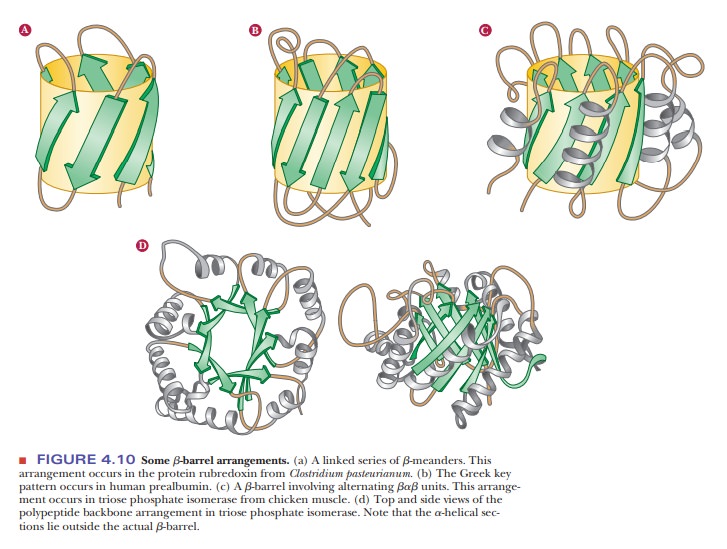
can often be found arranged into a β-barrel in the tertiary structure of the
protein (Figure 4.10). Motifs are important and tell us much about the folding
of proteins. However, these motifs do not allow us to predict anything about
the biological function of the protein because they are found in proteins and
enzymes with very dissimilar functions.

Many proteins that have the same type of function have similar protein sequences; consequently, domains with similar conformations are associated with the particular function. Many types of domains have been identified, including three different types of domains by which proteins bind to DNA. In addition, short polypeptide sequences within a protein direct the posttransla-tional modification and subcellular localization. For example, several sequences play a role in the formation of glycoproteins (ones that contain sugars in addi-tion to the polypeptide chain). Other specific sequences indicate that a protein is to be bound to a membrane or secreted from the cell. Still other specific sequences mark a protein for phosphorylation by a specific enzyme.
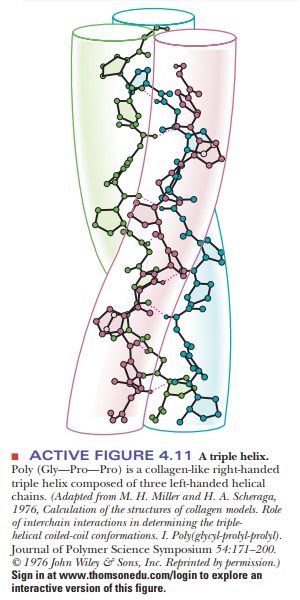
The Collagen Triple Helix
Collagen, a component of bone and connective tissue, is the most abundant protein in vertebrates. It is organized in water-insoluble fibers of great strength.
A collagen fiber consists of three polypeptide chains wrapped around each other in a ropelike twist, or triple helix. Each of the three chains has, within limits, a repeating sequence of three amino acid residues, X-Pro-Gly or X-Hyp-Gly, where Hyp stands for hydroxyproline, and any amino acid can occupy the first position, designated by X.
Proline
and hydroxyproline can constitute up to 30% of the residues in collagen.
Hydroxyproline is formed from proline by a specific hydroxylating enzyme after
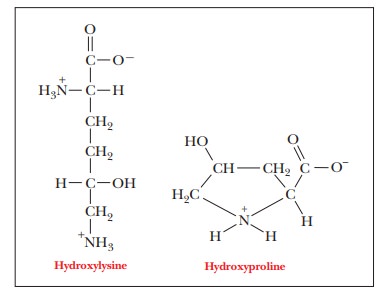
the amino acids are linked together. Hydroxylysine also occurs in collagen. In
the amino acid sequence of collagen, every third position must be occupied by
glycine. The triple helix is arranged so that every third residue on each chain
is inside the helix. Only glycine is small enough to fit into the space
available (Figure 4.11).
The three individual collagen chains are themselves helices that differ from the α-helix. They are twisted around each other in a superhelical arrangement toform a stiff rod. This triple helical molecule is called tropocollagen; it is 300 nm (3000 Å) long and 1.5 nm (15 Å) in diameter. The three strands are held together by hydrogen bonds involving the hydroxyproline and hydroxylysine residues.
The molecular
weight of the triple-stranded array is about 300,000; each strand contains
about 800 amino acid residues. Collagen is both intramolecularly and
intermolecularly linked by covalent bonds formed by reactions of lysine and
histidine residues. The amount of cross-linking in a tissue increases with age.
That is why meat from older animals is tougher than meat from younger animals.
Collagen in which the proline is not hydroxylated to hydroxyproline to the usual extent is less stable than normal collagen. Symptoms of scurvy, such as bleeding gums and skin discoloration, are the results of fragile collagen. The enzyme that hydroxylates proline and thus maintains the normal state of col-lagen requires ascorbic acid (vitamin C) to remain active. Scurvy is ultimately caused by a dietary deficiency of vitamin C.
Two Types of Protein Conformations: Fibrous and Globular
It is
difficult to draw a clear separation between secondary and tertiary structures.
The nature of the side chains in a protein (part of the tertiary structure) can
influence the folding of the backbone (the secondary structure). Comparing
collagen with silk and wool fibers can be illuminating. Silk fibers consist
largely of the protein fibroin, which, like collagen, has a fibrous structure,
but which, unlike collagen, consists largely of β-sheets. Fibers of wool
consist largely of the protein keratin, which is largely α-helical. The amino
acids of which collagen, fibroin, and keratin are composed determine which
conformation they will adopt, but all are fibrous
proteins (Figure 4.12a).
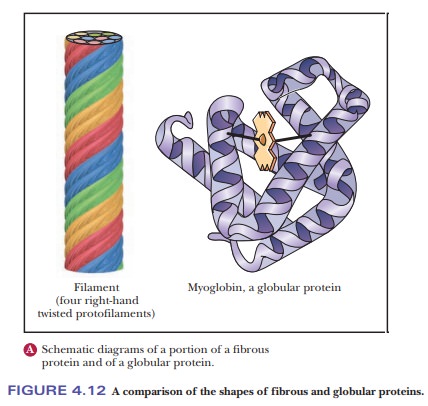
In other proteins, the backbone folds back on itself to produce a more or less spherical shape. These are called globular proteins (Figure 4.12b), and we shall see many examples of them.
Their helical and pleated-sheet sections can be
arranged so as to bring the ends of the sequence close to each other in three
dimensions. Globular proteins, unlike fibrous proteins, are water-soluble and
have compact structures; their tertiary and quaternary structures can be quite
complex.
Summary
Secondary structures are based on periodic
structures of the peptide backbone.
The most common secondary structures are the α-helix
and β-sheet.
Native proteins may have combinations of
various secondary structures
Regions of secondary structures can be combined
to form supersecondary structures, motifs, and domains.
One of
the most common structures is the triple helix of collagen, the protein that
makes up the bulk of connective tissue.
Related Topics