Chapter: Biochemistry: The Three-Dimensional Structure of Proteins
Protein Folding Dynamics
Protein Folding Dynamics
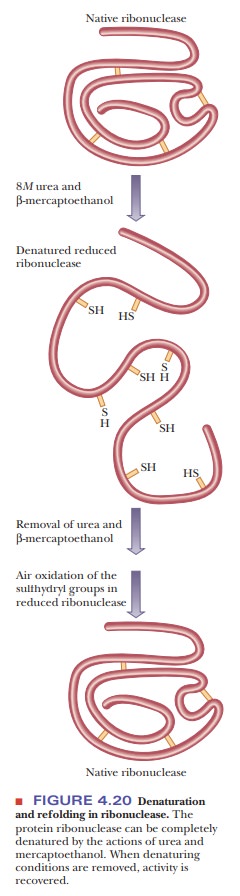
We know that the sequence of amino acids ultimately determines the three-dimensional structure of a protein. We also know that proteins can spontaneously adopt their native conformations, be denatured, and be renatured back into their native conformations, as was shown in Figure 4.20. These facts can lead us to the following question:
![]()
![]()
Can we predict the tertiary structure of a protein if we know its amino acid sequence?
With
modern computing techniques, we are able to predict protein structure. This is
becoming more and more possible as more powerful computers allow the processing
of large amounts of information. The encounter of biochemistry and computing
has given rise to the burgeoning field of bioinformatics.
Prediction of protein structure is one of the principal applications of
bioinformatics. Another important application is the comparison of base
sequences in nucleic acids, a topic we shall discuss, along with other methods
for working with nucleic acids. As we shall see, we can now predict protein
structure and function by knowing the nucleotide sequence of the gene that eventually
leads to the final protein.
The
first step in predicting protein architecture is a search of databases of known
structures for sequence homology
between the protein whose structure is to be determined and proteins of known
architecture, where the term homology
refers to similarity of two or more sequences. If the sequence of the known
pro-tein is similar enough to that of the protein being studied, the known
protein’s structure becomes the point of departure for comparative modeling. Use of mod-eling algorithms that compare the
protein being studied with known structures leads to a structure prediction.
This method is most useful when the sequence homology is greater than 25%–30%.
If the sequence homology is less than 25%–30%, other approaches are more
useful. Fold recognition algorithms
allow comparison with known folding motifs common to many secondary structures.
Here is an application of that information. Yet another method is de novo prediction, based on first
principles from chemistry, biology, and physics. This method too can give rise
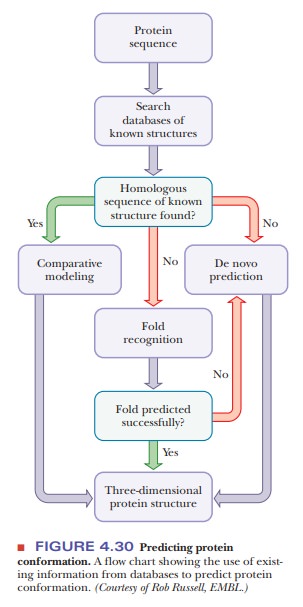
to struc-tures subsequently confirmed by X-ray crystallography. The flow chart
in Figure 4.30 shows how prediction techniques use existing information from
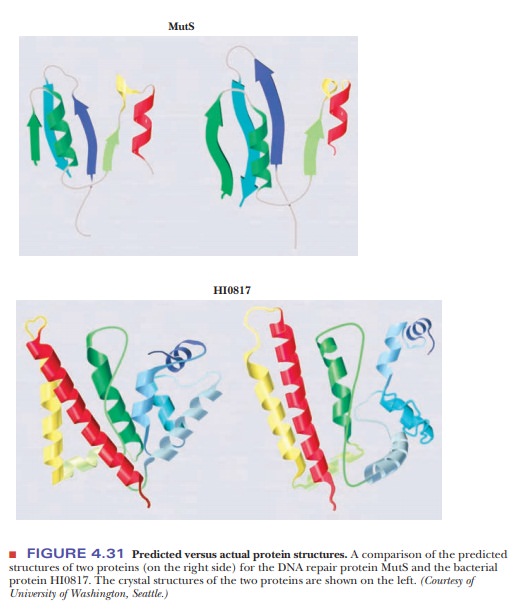
databases. Figure 4.31 shows a comparison of the predicted structures of two
proteins (on the right side) for the DNA repair protein MutS and the bacterial
protein HI0817. The crystal structures of the two proteins are shown on the
left.
Hydrophobic Interactions:A Case Study in Thermodynamics
Hydrophobic
interactions have important consequences in biochemistry and play a major role
in protein folding. Large arrays of molecules can take on definite structures
as a result of hydrophobic interactions. We have already seen the way in which
phospholipid bilayers can form one such array. Recall that phospholipids are molecules that have
polar head groups and long nonpolar tails of hydrocarbon chains. These bilayers
are less complex than a folded protein, but the interactions that lead to their
formation also play a vital role in protein folding. Under suitable conditions,
a double-layer arrangement is formed so that the polar head groups of many
molecules face the aqueous environment, while the nonpolar tails are in contact
with each other and are kept away from the aqueous environment. These bilayers
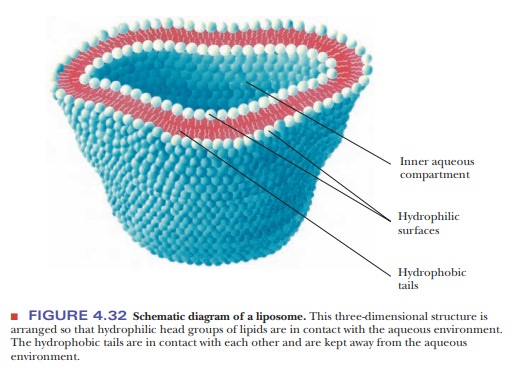
form three-dimensional structures called liposomes
(Figure 4.32). Such structures are useful model systems for biological
membranes, which consist of similar bilayers with proteins embedded in them.
The interactions between the bilayer and the embedded proteins are also
examples of hydrophobic interactions. The very existence of membranes depends
on hydrophobic interactions. The same hydrophobic interactions play a crucial
role in protein folding.

Hydrophobic
interactions are a major factor in the folding of proteins into the specific
three-dimensional structures required for their functioning as enzymes, oxygen
carriers, or structural elements. It is known experimentally that proteins tend
to be folded so that the nonpolar hydrophobic side chains are sequestered from
water in the interior of the protein, while the polar hydro-philic side chains
lie on the exterior of the molecule and are accessible to the aqueous
environment (Figure 4.33).
What makes hydrophobic interactions favorable?
Hydrophobic
interactions are spontaneous processes. The entropy of the Universe increases
when hydrophobic interactions occur.
∆Suniv > 0
As an
example, let us assume that we have tried to mix the liquid hydrocarbon hexane
(C6H14) with water and have obtained not a solution but a two-layer
system, one layer of hexane and one of water. Formation of a mixed solution is
nonspontaneous, and the formation of two layers is spontaneous. Unfavorable
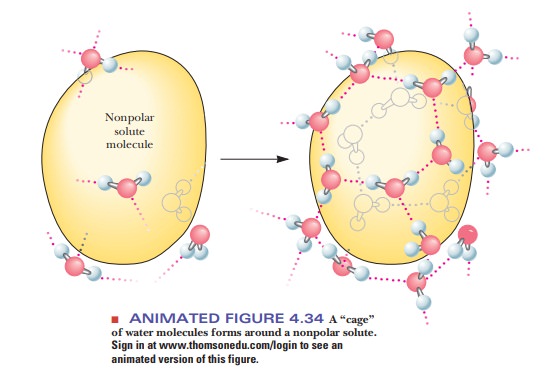
entropy terms enter into the picture if solution formation requires the
creation of ordered arrays of solvent, in this case water (Figure 4.34). The
water molecules surrounding the nonpolar molecules can hydrogen bond with each
other, but they have fewer possible orientations than if they were surrounded
by other water molecules on all sides. This introduces a higher degree of
order, preventing the dispersion of energy, more like the lattice of ice than
liquid water, and thus a lower entropy. The required entropy decrease is too
large for the process to take place. Therefore, nonpolar substances do not
dissolve in water; rather, nonpolar molecules associate with one another by
hydrophobic interactions and are excluded from water.
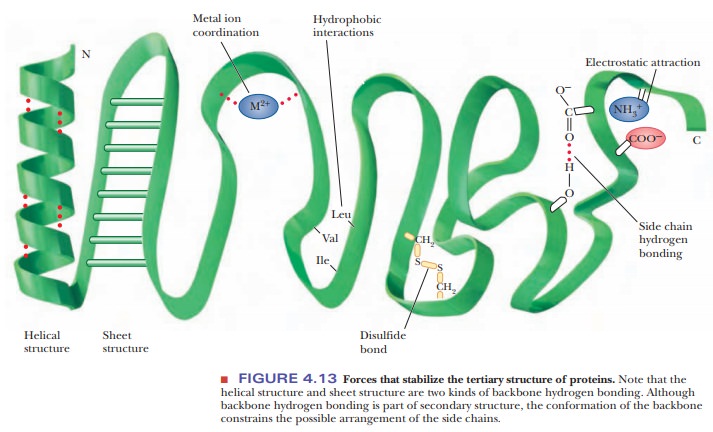
Many people think of hydrophobic interactions between amino acids back-ward. For example, if we look at Figure 4.13 and see the indication of hydro-phobic interactions between leucine, valine, and isoleucine, we might conclude that hydrophobic interactions refer to an attraction for these amino acids for each other. However, we now know that in reality it is not so much the attrac-tion of the nonpolar amino acids for each other, but rather it is more that they are forced together so that water can avoid having to interact with them.
The Importance of Correct Folding
The
primary structure conveys all the information necessary to produce the correct
tertiary structure, but the folding process in vivo can be a bit trickier. In
the protein-dense environment of the cell, proteins may begin to fold
incorrectly as they are produced, or they may begin to associate with other
proteins before completing their folding process. In eukaryotes, proteins may
need to remain unfolded long enough to be transported across the membrane of a
subcellular organelle.
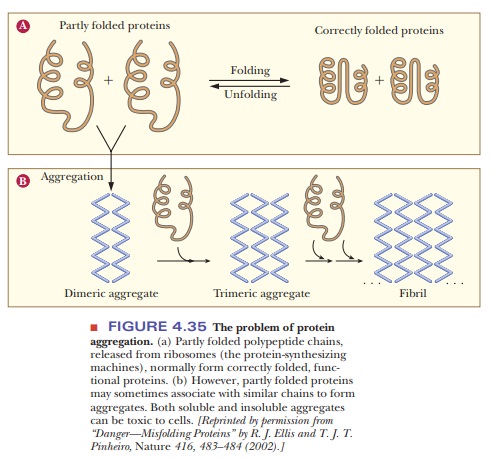
Correctly
folded proteins are usually soluble in the aqueous cell environ-ment, or they
are correctly attached to membranes. However, when proteins do not fold
correctly, they may interact with other proteins and form aggre-gates as shown
in Figure 4.35. This occurs because hydrophobic regions that should be buried
inside the protein remain exposed and interact with other hydrophobic regions
on other molecules. Several neurodegenerative disor-ders, such as Alzheimer’s,
Parkinson’s, and Huntington’s diseases are caused by accumulation of protein
deposits from such aggregates. See the following Biochemical Connections box
for a description of a deadly disease caused by protein misfolding.
Protein-Folding Chaperones
To help
avoid the protein misfolding problem, special proteins called chaperones aid in
the correct and timely folding of many proteins. The first such proteins
discovered were a family called hsp70 (for 70,000
MW heat-shock protein), which are proteins produced in E. coli grown above optimal temperatures. Chaperones exist in
organisms from prokaryotes through humans, and their mechanisms of action are
currently being studied. It is becoming more and more evident that protein
folding dynamics are crucial to protein function in vivo.
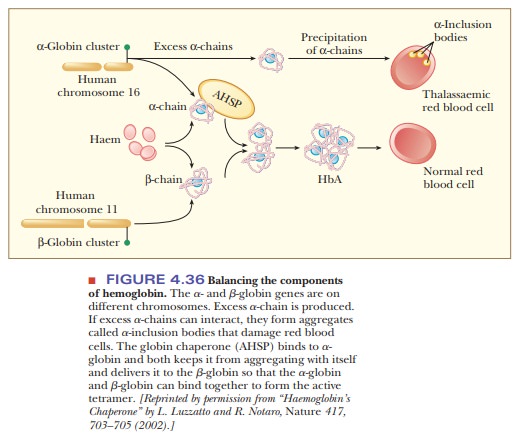
In the
blood, hemoglobin accumulates to a level of 340 g/L, which is a very large
amount of a single protein. The control of globin gene expression is
complicated and made more so by the fact that there are separate genes for the
α-chain and the β-chain, and they are found on different chromosomes. There are
also two α-globin genes for every β-globin gene, so there is always an excess
of the α-chain. Excess α-chains can form aggregates as shown in Figure 4.36,
which could lead to damaged red blood cells and a disease called thalas-semia. Thea-chains can also form
aggregates among themselves, leading to auseless form of hemoglobin. The secret
to success for hemoglobin production is to maintain the proper stoichiometry
between the two types of globin chains. The α-chains must be kept from
aggregating together so that there will be enough α-chain to complex with the
β-chain. In this way the α-chains will be occupied with β-chains and will not
form α-chain aggregates. Fortunately, there is a specific chaperone for the α-chain,
called α-hemoglobin stabilizing protein (AHSP). This chaperone prevents the α-chains
from causing the damage to blood cells as well as delivering them to the β-chains.
Protein folding is a very hot topic in biochemistry today. The following Biochemical Connections box describes a particularly striking example of the importance of protein folding.
Summary
Using the power of computers, we can now predict the tertiary
structure of a protein if we know its amino acid sequence.
A great deal of information regarding protein structure and
sequences can be found on the World Wide Web.
Chaperones are proteins that help another protein attain the
correct native conformation.
There is a specific chaperone for the formation of hemoglobin.
Protein
folding is critical to the proper function of a protein. There are diseases
caused by misfolded proteins. One of the most infamous is a dis-ease caused by
a misfolded protein called a prion. Misfolded prions cause spongiform
encephalopathy, otherwise known as mad-cow disease in the dairy industry or
Creutzfeldt-Jakob disease in humans.
Related Topics