Chapter: Medical Surgical Nursing: Assessment and Management of Patients With Hepatic Disorders
Management of Patients With Nonviral Hepatic Disorders
Management of Patients With Nonviral Hepatic Disorders
Certain
chemicals have toxic effects on the liver and when taken by mouth, inhaled, or
injected parenterally produce acute liver cell necrosis, or toxic hepatitis.
The chemicals most commonly implicated in this disease are carbon
tetrachloride, phosphorus, chloroform, and gold compounds. These substances are
true he-patotoxins. Many medications may induce hepatitis but are sen-sitizing
rather than toxic. The result, drug-induced hepatitis, is similar to acute
viral hepatitis, but parenchymal destruction tends to be more extensive. Some
medications that can lead to hepati-tis are isoniazid, halothane,
acetaminophen, and certain antibi-otics, antimetabolites, and anesthetic
agents.
TOXIC HEPATITIS
Toxic
hepatitis resembles viral hepatitis in onset. Obtaining a history of exposure
to hepatotoxic chemicals, medications, or other agents assists in early
treatment and removal of the of-fending agent. Anorexia, nausea, and vomiting
are the usual symptoms; jaundice and hepatomegaly are noted on physical as-sessment.
Symptoms are more intense for the more severely toxic patient.
Recovery
from acute toxic hepatitis is rapid if the hepatotoxin is identified early and
removed or if exposure to the agent has been limited. Recovery is unlikely if
there is a prolonged period between exposure and onset of symptoms. There are
no effective antidotes. The fever rises; the patient becomes toxic and
pros-trated. Vomiting may be persistent, with the emesis containing blood.
Clotting abnormalities may be severe, and hemorrhages may appear under the
skin. The severe GI symptoms may lead to vascular collapse. Delirium, coma, and
seizures develop, and within a few days the patient may die of fulminant
hepatic fail-ure (discussed below) unless he or she receives a liver transplant.
Short
of liver transplantation, few treatment options are avail-able. Therapy is
directed toward restoring and maintaining fluid and electrolyte balance, blood
replacement, and comfort and sup-portive measures. A few patients recover from
acute toxic hepati-tis only to develop chronic liver disease. If the liver
heals, there may be scarring, followed by postnecrotic cirrhosis.
DRUG-INDUCED HEPATITIS
Drug-induced
hepatitis is responsible for 20% to 25% of cases of acute hepatic failure in
the United States (Maddrey, Schiff & Sorrell, 2001). Manifestations of
sensitivity to a medication may occur on the first day of its use or not until
several months later, depending on the medication. Usually the onset is abrupt,
with chills, fever, rash, pruritus, arthralgia, anorexia, and nausea. Later,
there may be jaundice and dark urine and an enlarged and tender liver. When the
offending medication is withdrawn, symptoms may gradually subside. However,
reactions may be severe and even fatal, even though the medication is stopped.
If fever, rash, or pruritus occurs from any medication, its use should be
stopped immediately.
Although
any medication can affect liver function, use of acet-aminophen (found in many
over-the counter medications used to treat fever and pain) has been identified
as the leading cause of acute liver failure (Ostapowicz, Fontana, Schiodt, et
al., 2002). Others commonly associated with liver injury include but are not
limited to anesthetic agents, medications used to treat rheumatic and musculoskeletal
disease, antidepressants, psychotropic med-ications, anticonvulsants, and
anti-tuberculosis agents.
Inhalational
agents of the halothane family (halokanes) are metabolized by the liver and
excreted in bile. These volatile anes-thetics may also decrease hepatic blood
flow. Halothane hepatitis is a dreaded but rare complication of halothane
administration. Sevoflurane and desflurane may have less hepatotoxic effects
than halokanes. Nitrous oxide, an adjunct to halokanes, is not hepa-totoxic.
Because it undergoes little hepatic metabolism, isoflurane is considered the
anesthetic agent of choice in patients with liver disease (Bacon & Di
Bisceglie, 2000).
Although
its efficacy is uncertain, a short course of high-dose corticosteroids may be
used in patients with severe hypersensitiv-ity. Liver transplantation is an
option for drug-induced hepatitis, but outcomes may not be as successful as
with other causes of liver failure.
FULMINANT HEPATIC FAILURE
Fulminant hepatic failure is the clinical syndrome
of suddenand severely impaired liver function in a previously healthy per-son.
According to the original and generally accepted definition, fulminant hepatic
failure develops within 8 weeks of the first symptoms of jaundice (Maddrey et
al., 2001). Patterns of the progression from jaundice to encephalopathy have
been identi-fied and have led to proposals of time-based classifications, but
no agreement as to these classifications has been reached. How-ever, three
categories are frequently cited: hyperacute, acute, and subacute liver failure.
In hyperacute liver failure, the duration of jaundice before the onset of
encephalopathy is 0 to 7 days; in acute liver failure, it is 8 to 28 days; and
in subacute liver failure, it is 28 to 72 days. The prognosis for fulminant
hepatic failure is much worse than for chronic liver failure. However, in
fulminant failure, the hepatic lesion is potentially reversible, with survival
rates of approximately 50% to 85% (survival rates depend greatly on the
etiology of liver failure). Those who do not survive die of massive
hepatocellular injury and necrosis (Maddrey et al., 2001).
Viral
hepatitis is a common cause of fulminant hepatic failure; other causes include
toxic medications (eg, acetaminophen) and chemicals (eg, carbon tetrachloride),
metabolic disturbances (Wil-son’s disease, a hereditary syndrome with
deposition of copper in the liver), and structural changes (Budd-Chiari
syndrome, an ob-struction to outflow in major hepatic veins).
Jaundice
and profound anorexia may be the initial reasons the patient seeks health care.
Fulminant hepatic failure is often accompanied by coagulation defects, renal
failure and elec-trolyte disturbances, infection, hypoglycemia, encephalopathy,
and cerebral edema.
Management
The
key to optimizing treatment is rapid recognition of acute liver failure and
intensive interventions. Treatment modalities may in-clude plasma exchanges
(plasmapheresis) or charcoal hemoper-fusion for the removal (theoretically) of
potentially harmful metabolites (Kaptanoglu & Blei, 2000); however, more
clinical trials are needed to determine their effects or outcomes. Hepato-cytes
within synthetic fiber columns have been tested as liver sup-port systems
(liver assist devices) and a bridge to transplantation.
Research
into interventions for acute liver failure has begun to focus on techniques
that combine the efficacy of a whole liver with the convenience and
biocompatibility of hemodialysis. The acronyms ELAD (extracorporeal liver
assist devices) and BAL (bioartificial liver) have been used to describe these
hybrid devices. These temporary devices help patients to survive until
transplanta-tion is possible. The BAL exposes separated plasma to a cartridge
containing porcine liver cells after the plasma has flowed through a charcoal
column that removes substances toxic to hepatocytes. The ELAD device exposes
whole blood to cartridges containing human hepatoblastoma cells, resulting in
removal of toxic sub-stances. Similar extracorporeal circuits using xenografts will likely be studied in
the near future (Maddrey et al., 2001). Although these approaches appear
promising, controlled studies are required.
There
is a high risk for cerebral edema, a life-threatening complication, in patients
with fulminant liver failure with stage 4 encephalopathy. The cause is not
fully understood, although disruption of the blood–brain barrier and plasma
leaking into the cerebrospinal fluid has been proposed as one theory (Sherlock
& Dooley, 2002). These patients require intracranial pressure mon-itoring.
Measures to promote adequate cerebral perfusion include careful fluid balance
and hemodynamic assessments, a quiet en-vironment, and diuresis with mannitol,
an osmotic diuretic.
To
prevent surges in intracranial pressure related to agitation, barbiturate
anesthesia or pharmacologic paralysis and sedation are indicated. Other support
measures include monitoring for and treating hypoglycemia, coagulopathies, and
infection. Despite these treatment modalities, the mortality rate remains high.
Con-sequently, liver transplantation has become the treatment of choice for
fulminant hepatic failure.
HEPATIC CIRRHOSIS
Cirrhosis
is a chronic disease characterized by replacement of nor-mal liver tissue with
diffuse fibrosis that disrupts the structure and function of the liver. There
are three types of cirrhosis or scarring of the liver:
•
Alcoholic cirrhosis, in which the scar tissue
characteristi-cally surrounds the portal areas. This is most frequently due to
chronic alcoholism and is the most common type of cirrhosis.
•
Postnecrotic cirrhosis, in which there are broad
bands of scar tissue as a late result of a previous bout of acute viral
hepatitis.
•
Biliary cirrhosis, in which scarring occurs in the
liver around the bile ducts. This type usually is the result of chronic
bil-iary obstruction and infection (cholangitis); it is much less common than
the other two types.
The
portion of the liver chiefly involved in cirrhosis consists of the portal and
the periportal spaces, where the bile canaliculi of each lobule communicate to
form the liver bile ducts. These areas become the sites of inflammation, and
the bile ducts be-come occluded with inspissated (thickened) bile and pus. The
liver attempts to form new bile channels; hence, there is an over-growth of
tissue made up largely of disconnected, newly formed bile ducts and surrounded
by scar tissue.
Clinical
manifestations include intermittent jaundice and fever. Initially the liver is
enlarged, hard, and irregular, but even-tually it atrophies.
Pathophysiology
Although
several factors have been implicated in the etiology of cirrhosis, alcohol
consumption is considered the major causative factor. Cirrhosis occurs with
greatest frequency among alcoholics. Although nutritional deficiency with
reduced protein intake con-tributes to liver destruction in cirrhosis,
excessive alcohol intake is the major causative factor in fatty liver and its
consequences. Cirrhosis, however, has also occurred in people who do not
con-sume alcohol and in those who consume a normal diet and have a high alcohol
intake.
Some
people appear to be more susceptible than others to this disease, whether or
not they are alcoholics or malnourished. Other factors may play a role,
including exposure to certain chemicals (carbon tetrachloride, chlorinated
naphthalene, arsenic, or phos-phorus) or infectious schistosomiasis. Twice as
many men as women are affected, although women are at greater risk of
devel-oping alcohol-induced liver disease for an as yet undiscovered reason.
Most patients are between 40 and 60 years of age. Each year more than 25,000
people die of chronic liver diseases and cirrhosis in the United States (NIDDK,
2000).
Alcoholic
cirrhosis is characterized by episodes of necrosis in-volving the liver cells,
sometimes occurring repeatedly throughout the course of the disease. The
destroyed liver cells are replaced grad-ually by scar tissue; eventually the
amount of scar tissue exceeds that of the functioning liver tissue. Islands of
residual normal tissue and regenerating liver tissue may project from the
constricted areas, giv-ing the cirrhotic liver its characteristic hobnail
appearance. The dis-ease usually has an insidious onset and a protracted
course, occasionally proceeding over a period of 30 or more years.
The
prognosis of different forms of cirrhosis caused by vari-ous liver diseases has
been investigated in several studies. Of the many prognostic indicators, the
Child’s classification seems most useful in predicting the outcome of patients
with liver disease (Table 39-5). It is also used in choosing management
approaches.
Clinical Manifestations
Signs
and symptoms of cirrhosis increase in severity as the disease progresses. The
severity of the manifestations helps to categorize the disorder into two main
presentations (Chart 39-10).
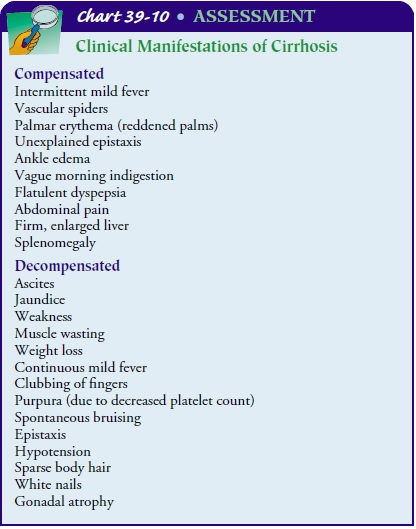
Compensated
cirrhosis, with its less severe, often vague symp-toms, may be discovered
secondarily at a routine physical exam-ination. The hallmarks of decompensated
cirrhosis result from failure of the liver to synthesize proteins, clotting
factors, and other substances and manifestations of portal hypertension.
LIVER ENLARGEMENT
Early
in the course of cirrhosis, the liver tends to be large and its cells loaded
with fat. The liver is firm and has a sharp edge no-ticeable on palpation.
Abdominal pain may be present because of recent, rapid enlargement of the
liver, producing tension on the fibrous covering of the liver (Glisson’s
capsule). Later in the dis-ease, the liver decreases in size as scar tissue
contracts the liver tis-sue. The liver edge, if palpable, is nodular.
PORTAL OBSTRUCTION AND ASCITES
These
late manifestations are due partly to chronic failure of liver function and
partly to obstruction of the portal circula-tion. Practically all the blood
from the digestive organs is col-lected in the portal veins and carried to the
liver. Because a cirrhotic liver does not allow the blood free passage, it
backs up into the spleen and the GI tract and these organs become the seat of
chronic passive congestion; that is, they are stagnant with blood and thus
cannot function properly. Indigestion and altered bowel function result. Fluid
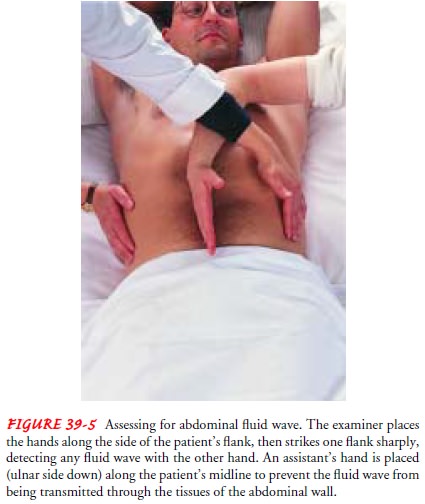
rich in protein may accu-mulate in the peritoneal cavity, producing ascites.
This can be demonstrated through percussion for shifting dullness or a fluid
wave (see Fig. 39-5).
INFECTION AND PERITONITIS
Bacterial
peritonitis may develop in cirrhotic patients with as-cites in the absence of
an intra-abdominal source of infection or an abscess. This condition is
referred to as spontaneous bacte-rial peritonitis. Bacteremia is believed to be
the most likely route of infection. Clinical signs may be absent; paracentesis
may be necessary for diagnosis. Antibiotic therapy is effective in the
treatment and prevention of recurrent episodes of spontaneous bacterial
peritonitis.
GASTROINTESTINAL VARICES
The
obstruction to blood flow through the liver resulting from the fibrotic changes
also results in the formation of collateral blood ves-sels in the GI system and
shunting of blood from the portal vessels into blood vessels with lower
pressures. As a result, the patient with cirrhosis often has prominent,
distended abdominal blood vessels, which are visible on abdominal inspection
(caput medusae), and distended blood vessels throughout the GI tract. The
esophagus, stomach, and lower rectum are common sites of collateral blood
vessels. These distended blood vessels form varices or hemorrhoids, depending
on their location (see Fig. 39-6).
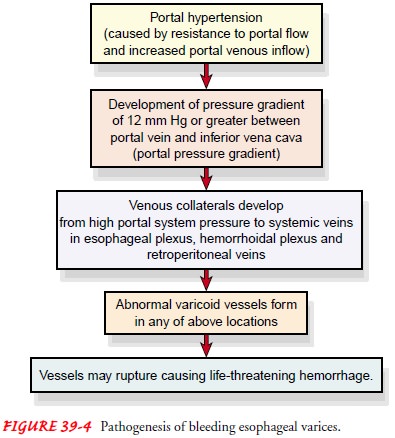
Because
these vessels were not intended to carry the high pres-sure and volume of blood
imposed by cirrhosis, they may rup-ture and bleed. Therefore, assessment must
include observation for occult and frank bleeding from the GI tract.
Approximately 25% of patients develop minor hematemesis; others have pro-fuse
hemorrhage from gastric and esophageal varices (Bacon & Di Bisceglie,
2000).
EDEMA
Another
late symptom of cirrhosis is edema, which is attributed to chronic liver
failure. A reduced plasma albumin concentration predisposes the patient to the
formation of edema. Edema is gen-eralized but often affects lower extremities,
upper extremities, and the presacral area. Facial edema is not typical.
Overproduc-tion of aldosterone occurs, causing sodium and water retention and
potassium excretion.
VITAMIN DEFICIENCY AND ANEMIA
Because
of inadequate formation, use, and storage of certain vit-amins (notably
vitamins A, C, and K), signs of their deficiency are common, particularly
hemorrhagic phenomena associated with vitamin K deficiency. Chronic gastritis
and impaired GI function, together with inadequate dietary intake and impaired
liver function, account for the anemia often associated with cir-rhosis. The
anemia and the patient’s poor nutritional status and poor state of health
result in severe fatigue, which interferes with the ability to carry out
routine daily activities.
MENTAL DETERIORATION
Additional
clinical manifestations include deterioration of men-tal function with
impending hepatic encephalopathy and hepatic coma. Neurologic assessment is
indicated and includes the pa-tient’s general behavior, cognitive abilities,
orientation to time and place, and speech patterns.
Assessment and Diagnostic Findings
The
extent of liver disease and the type of treatment are deter-mined after
reviewing the laboratory findings. Because the func-tions of the liver are
complex, there are many diagnostic tests that may provide information about
liver function (see Table 39-1). The patient needs to know why these tests are
being performed and ways to cooperate.

In
severe parenchymal liver dysfunction, the serum albumin level tends to decrease
and the serum globulin level rises. Enzyme tests indicate liver cell damage:
serum alkaline phosphatase, AST, ALT, and GGT levels increase, and the serum
cholinesterase level may decrease. Bilirubin tests are performed to measure
bile ex-cretion or bile retention; elevated levels can occur with cirrhosis and
other liver disorders. Prothrombin time is prolonged.
Ultrasound
scanning is used to measure the difference in density of parenchymal cells and
scar tissue. CT, MRI, and ra-dioisotope liver scans give information about
liver size and he-patic blood flow and obstruction. Diagnosis is confirmed by
liver biopsy. Arterial blood gas analysis may reveal a ventilation– perfusion
imbalance and hypoxia.
Medical Management
The
management of the patient with cirrhosis is usually based on the presenting
symptoms. For example, antacids are prescribed to decrease gastric distress and
minimize the possibility of GI bleed-ing. Vitamins and nutritional supplements
promote healing of damaged liver cells and improve the general nutritional
status. Potassium-sparing diuretics (spironolactone [Aldactone], tri-amterene
[Dyrenium]) may be indicated to decrease ascites, if present; these diuretics
are preferable to other diuretic agents be-cause they minimize the fluid and
electrolyte changes common with other agents. An adequate diet and avoidance of
alcohol are essential. Although the fibrosis of the cirrhotic liver cannot be
re-versed, its progression may be halted or slowed by such measures.
Preliminary
studies indicate that colchicine, an anti-inflammatory agent used to treat the
symptoms of gout, may in-crease the length of survival in patients with mild to
moderate cirrhosis. Improved survival has been observed in patients with
alcoholic cirrhosis. Colchicine is believed to reverse the fibrotic processes
in cirrhosis, and this has improved survival (Bacon & Di Bisceglie, 2000).
Related Topics