Chapter: Clinical Anesthesiology: Regional Anesthesia & Pain Management: Chronic Pain Management
Interventional Therapies: Pharmacological Interventions
Interventional Therapies
Interventional pain therapy may take the form of pharmacological
treatment, nerve blocks with local anesthetics and steroid or a neurolytic
solution, radiofrequency ablation, neuromodulatory tech-niques, or multidisciplinary
treatment (psychologi-cal interventions, physical or occupational therapy, or
modalities such as acupuncture).
PHARMACOLOGICAL INTERVENTIONS
Pharmacological interventions in pain management include acetaminophen,
cyclooxygenase (COX) inhibitors, opioids, antidepressants, neurolepticagents,
anticonvulsants, corticosteroids, and sys-temic administration of local
anesthetics.
Acetaminophen
Acetaminophen (paracetamol) is an oral analge-sic and antipyretic agent
that recently has become available in the United States as an intravenous
preparation (Ofirmev) for inpatient use. It inhibits prostaglandin synthesis
but lacks significant antiin-flammatory activity. Acetaminophen has few side
effects but is hepatotoxic at high doses. The recom-mended adult maximum daily
limit is 3000 mg/d, reduced from a previously recommended limit of 4000 mg/d.
Isoniazid, zidovudine, and barbiturates can potentiate acetaminophen toxicity.
Nonsteroidal Antiinflammatory Drugs (NSAIDs)
Nonopioid oral analgesics include salicylates, acetaminophen, and NSAIDs
(see Table 47–11). NSAIDs inhibit prostaglandin synthesis (COX).
Prostaglandins sensitize and amplify
nocicep-tive input, and blockade of their synthesis results in the analgesic,
antipyretic, and antiinflamma-tory properties characteristic of NSAIDs. At
least two types of COX are recognized. COX-1 is con-stitutive and widespread
throughout the body, but COX-2 is expressed primarily with inflammation. Some
types of pain, particularly pain that follows orthopedic and gynecological
surgery, respond very well to COX inhibitors. COX inhibitors likely have
important peripheral and central nervous system actions. Their analgesic action
is limited by side effects and toxicity at higher doses. Selective COX-2
inhibitors, such as celecoxib, appear to have lower toxicity, particularly
gastrointestinal side effects. Moreover, COX-2 inhibitors do not interfere with
platelet aggregation. The COX-2 inhibitor rofecoxib increases the risk of
cardiovascular complications; as a result, it has been taken off of the market
in the United States.
All of the nonopioid oral analgesic agents
are well absorbed enterally. Food delays absorption but otherwise has no effect
on bioavailability. Because most of these agents are highly protein bound (>80%), they can displace
other highly bound drugs such as warfarin. All undergo hepatic metabolism and
are renally excreted. Dosages should therefore be reduced, or alternative
medications selected, in patients with hepatic or renal impairment.
The most common side effects of aspirin (ace-tylsalicylic acid, ASA) and other NSAIDs are stom-ach upset, heartburn, nausea, and dyspepsia; some patients develop ulceration of the gastric mucosa, which appears to be due to inhibition of prosta-glandin-mediated mucus and bicarbonate secretion. Diclofenac is available as both an oral preparation and a topical gel or patch that may be less likely to contribute to gastric distress.
Other side effects of NSAIDs include
dizziness, headache, and drowsiness. With the exception of selective COX-2
inhibitors, all other COX inhibi-tors induce platelet dysfunction. Aspirin
irreversibly acetylates platelets, inhibiting platelet adhesiveness for 1–2
weeks, whereas the antiplatelet effect of other NSAIDs is reversible and lasts
approximately five elimination half-lives (24–96 h). This antiplate-let effect
does not appear to appreciably increase the incidence of postoperative
hemorrhage follow-ing most outpatient procedures. NSAIDs can exac-erbate
bronchospasm in patients with the triad of nasal polyps, rhinitis, and asthma.
ASA should not be used in children with varicella or influenza infec-tions
because it may precipitate Reye’s syndrome. Lastly, NSAIDs can cause acute
renal insufficiency and renal papillary necrosis, particularly in patients with
underlying renal dysfunction.
Antidepressants
Antidepressants are most useful for patients
with neuropathic pain. These medicationsdemonstrate an analgesic effect that
occurs at a dose lower than that needed for antidepressant activity,and both
of these actions are due to blockade of pre-synaptic reuptake of serotonin,
norepinephrine, or both. Older tricyclic agents appear to be more effec-tive
analgesics than selective serotonin reuptake inhibitors (SSRIs). Serotonin and
norepinephrine reuptake inhibitors (SNRIs) may provide the most favorable
balance between analgesic efficacy and side effects. Antidepressants potentiate
the action of opioids and frequently help normalize sleep patterns.
All antidepressant medications undergo exten-sive first-pass hepatic
metabolism and are highly protein bound. Most are highly lipophilic and have
large volumes of distribution. Elimination half-lives of most of these
medications vary between 1 and 4 days, and many have active metabolites.
Available agents differ in their side effects (see Table 47–13), which include
antimuscarinic effects (dry mouth, impaired visual accommodation, urinary
retention, and constipation), antihistaminic effects (sedation and increased gastric
pH), α-adrenergic blockade
(orthostatic hypotension), and a quinidine-like effect (atrioventricular block,
QT prolongation, tor-sades de pointes).
Serotonin & Norepinephrine Reuptake Inhibitors (SNRIs)
Milnacipran, along with the SNRI duloxetine
and the anticonvulsant pregabalin, has also been approved in the United States
by the FDA for the treatment of fibromyalgia. It has an elimination half-life
of 8 h, is minimally metabolized by the liver, and is primarily excreted
unchanged in the urine.
Duloxetine (Cymbalta) is useful in the treat-ment of neuropathic pain,
depression, and fibromy-algia. It has a half-life of 12 h, is metabolized by
the liver, and most of its metabolites are excreted in the urine.
Absolute and relative contraindications for
the use of SNRIs include known hypersensitivity, usage of other drugs that act
on the central nervous system (including monoamine oxidase inhibitors), hepatic
and renal impairment, uncontrolled narrow-angle glaucoma, and suicidal
ideation. Common side effects include nausea, headache, dizziness,
consti-pation, insomnia, hyperhydrosis, hot flashes, vomit-ing, palpitations,
dry mouth, and hypertension.
Neuroleptics
Neuroleptic medications may occasionally be use-ful for patients with
refractory neuropathic pain, and may be most helpful in patients with marked
agitation or psychotic symptoms. The most com-monly used agents are
fluphenazine, haloperi-dol, chlorpromazine, and perphenazine. Their therapeutic
action appears to be due to blockade of dopaminergic receptors in mesolimbic
sites. Unfortunately, the same action in nigrostriatal pathways can produce
undesirable extrapyramidal side effects, such as masklike facies, a festinating
gait, cogwheel rigidity, and bradykinesia. Some patients also develop acute dystonic
reactions such as oculogyric crisis and torticollis. Long-term side effects
include akathisia (extreme restlessness) and tardive dyskinesia (involuntary
choreoathetoid movements of the tongue, lip smacking, and trun-cal
instability). Like antidepressants, many of these drugs also have
antihistaminic, antimuscarinic, and α-adrenergic–blocking
effects.
Antispasmodics & Muscle Relaxants
Antispasmodics may be helpful for patients
with musculoskeletal sprain and pain associated with spasm or contractures.
Tizanidine (Zanaflex) is a centrally acting α2-adrenergic agonist used in the treatment of
muscle spasm in conditions such as multiple sclerosis, low back pain, and
spastic diple-gia. Cyclobenzaprine (Flexeril) also may be effective for these
conditions. Its precise mechanism of action is unknown.
Baclofen (Gablofen, Lioresal), a GABA B ago-nist, is particularly effective in the
treatment of muscle spasm associated with multiple sclerosis or spinal cord
injury when administered by continuous intrathecal drug infusion. Abrupt
discontinuation of this medication has been associated with fever, altered
mental status, pronounced muscle spasticity or rigidity, rhabdomyolysis, and
death.
Corticosteroids
Glucocorticoids are extensively used in pain
man-agement for their antiinflammatory and possi-bly analgesic actions. They
may be given topically, orally, or parenterally (intravenously,
subcutane-ously, intrabursally, intraarticularly, or epidurally). Table
47–15 lists the most commonly used agents,which
differ in potency, relative glucocorticoid and mineralocorticoid activities,
and duration or action. Large doses or prolonged administration result in
significant side effects. Excess glucocorticoid activity can produce
hypertension, hyperglycemia, increased susceptibility to infection, peptic
ulcers, osteoporosis, aseptic necrosis of the femoral head, proximal myopathy,
cataracts, and, rarely, psychosis. Patients with diabetes may have elevated
blood glu-cose levels after corticosteroid injections. Patients can also
develop the physical features characteristic of Cushing’s syndrome. Excess
mineralocorticoid

activity causes sodium retention and hypokalemia, and can precipitate
congestive heart failure.
Many corticosteroid preparations are suspen-sions, rather than
solutions, and the relative par-ticulate size of a given glucocorticoid
suspension may affect the risk of neural damage due to arterial occlusion when
accidental arterial injection occurs. Because of the relatively small size of
its suspension particles, dexamethasone is becoming the preferred
corticosteroid for injection procedures involving relatively vascular areas,
such as the head and neck region.
Anticonvulsants
Anticonvulsant medications are useful for
patients with neuropathic pain, especially trigeminal neuralgia and diabetic
neuropathy. These agents block voltage-gated calcium or sodium chan-nels and
can suppress the spontaneous neural dis-charges that play a major role in these
disorders. The most commonly utilized agents are phenytoin (Dilantin),
carbamazepine (Tegretol), valproic acid (Depakene, Stavzor), clonazepam
(Klonopin), and gabapentin (Neurontin) (Table 47–14). Pregabalin (Lyrica) is a
newer agent that has been approved for the treatment of diabetic peripheral
neuropathy and fibromyalgia but is widely prescribed for all forms of
neuropathic pain. Lamotrigine (Lamictal) and topiramate (Topamax) may also be
effective. All are highly protein bound and have relatively long half-lives.
Carbamazepine (Carbatrol, Equetro, Tegretol) has a slow and unpredictable
absorption, which requires monitoring of blood levels for optimal effi-cacy.
Phenytoin may be effective, but there is a pos-sible side effect of gum
hyperplasia. Levetiracetam (Keppra) and oxcarbazepine (Trileptal) have been
used as adjuvant pain therapies. Gabapentin and pregabalin may also be
effective adjuvants for the treatment of acute postoperative pain.
Local Anesthetics
Systemic infusion of local anesthetic
medication produces sedation and central analgesia and is occa-sionally used in
the treatment of patients with neuro-pathic pain. The resultant analgesia may
outlast the pharmacokinetic profile of the local anesthetic and break the “pain
cycle.” Lidocaine, procaine, and chlo-roprocaine are the most commonly used
agents. They are given either as a slow bolus or by continuous infu-sion.
Lidocaine is given by infusion over 5–30 min for a total of 1–5 mg/kg.
Procaine, 200–400 mg, can be given intravenously over the course of 1–2 h,
whereas chloroprocaine (1% solution) is infused at a rate of 1 mg/kg/min for a total
of 10–20 mg/kg. Monitoring by qualified medical personnel should include
electrocardiographic data, blood pressure, respiration, pulse oximetry, and
mental status, and full resuscitation equipment should be immediately
available. Signs of toxicity, such as tinnitus, slurring of speech, excessive
sedation, or nystagmus, neces-sitate slowing or discontinuing the infusion to
avoid the progression to seizures.
Patients who do not respond satisfactorily to anticonvulsants but
respond to intravenous local anesthetics may benefit from chronic oral
antiar-rhythmic therapy. Mexiletine (150–300 mg every 6–8 h) is a class 1B
antiarrhythmic that is commonly used and generally well tolerated.
A 5% lidocaine transdermal patch (Lidoderm) containing 700 mg of
lidocaine has been approved for the treatment of PHN. One to three patches may
be applied to dry, intact skin, alternating 12 h on, then 12 h off. Topical
lidocaine preparations, in con-centrations up to 5%, may be helpful in the
treat-ment of some neuropathic pain conditions.
α2-Adrenergic Agonists
The primary effect of α2-adrenergic
agonists is acti-vation of descending inhibitory pathways in the dorsal horn.
Epidural and intrathecal α2-adrenergic
agonists are particularly effective in the treatment of neuropathic pain and
opioid tolerance. Cloni-dine (Catapres), a direct-acting α2-adrenergic ago-nist, is
effective as an adjunctive medication in the treatment of severe pain. When
administered orally, the dosage is 0.1–0.3 mg twice daily; a transdermal patch
(0.1–0.3 mg/d) is also available and is usually applied for 7 d. When used in
combination with a local anesthetic or opioid in an epidural or intra-thecal
infusion, clonidine may contribute to a syn-ergistic or prolonged analgesic
effect, especially for neuropathic pain.
Opioids
The most commonly prescribed oral opioid
agents are codeine, oxycodone, and hydrocodone. They areeasily absorbed, but
hepatic first-pass metabolism lim-its systemic delivery. Like other opioids,
they undergo hepatic biotransformation and conjugation before renal
elimination. Codeine is transformed by the liver into morphine. The side
effects of orally administered opioids are similar to those of systemic
opioids. When prescribed on a fixed schedule, stool softeners or laxa-tives are
often indicated. Tramadol (Rybix, Ryzolt, Ultram) is a synthetic oral opioid
that also blocks neu-ronal reuptake of norepinephrine and serotonin. It appears
to have the same efficacy as the combination of codeine and acetaminophen but,
unlike others, it is associated with significantly less respiratory depres-sion
and has little effect on gastric emptying.
Moderate to severe cancer pain is usually
treated with an immediate-release morphine preparation (eg, liquid morphine,
Roxanol, 10–30 mg every 1–4 h). These preparations have an effective half-life
of 2–4 h. Once the patient’s daily requirements are determined, the same dose
can be given in the form of a sustained-release morphine preparation (MS Contin
or Oramorph SR), which is dosed every 8–12 h. The immediate-release preparation
is then used only for breakthrough pain (PRN). Oral trans-mucosal fentanyl
lozenges (Actiq, 200–1600 mcg) can also be used for breakthrough pain.
Excessive sedation can be treated with dextroamphetamine (Dexedrine, ProCentra)
or methylphenidate (Rit-alin), 5 mg in the morning and 5 mg the early
after-noon. Most patients require a stool softener. Nausea may be treated with
transdermal scopolamine, oral meclizine, or metoclopramide. Hydromorphone
(Dilaudid) is an excellent alternative to morphine, particularly in elderly
patients (because of fewer side effects) and in patients with impaired renal
function. Methadone (Dolophine) is reported to have a half-life of 15–30 h, but
clinical duration is shorter and quite variable (usually 6–8 h).
Patients
who experience opioid
tolerance require escalating doses of opioid to main-tain the same analgesic
effect. Physical dependencemanifests in opioid withdrawal when
the opioid medication is either abruptly discontinued or the dose is abruptly
and significantly decreased. Psy-chological dependence, characterized by
behavioral changes focusing on drug craving, is rare in
can-cer patients. The development of opioid tolerance is highly variable but
results in some desirable effects such as decreased opioid-related sedation,
nausea, and respiratory depression. Unfortunately, many patients continue to
suffer from constipation. Physical dependence occurs in all patients receiving
large doses of opioids for extended periods. Opioid withdrawal phenomena can be
precipitated by the administration of opioid antagonists. Future con-comitant
use of peripheral opioid antagonists that do not cross the blood–brain barrier,
such as meth-ylnaltrexone (Relistor) and alvimopan (Entereg), may help reduce
systemic side effects without sig-nificantly affecting analgesia.
Tapentadol (Nucynta), a μ-opioid receptor
ago-nist that also has norepinephrine reuptake inhibi-tion properties, has
recently been introduced for the management of acute and chronic pain. This
opioid may be associated with less nausea and vomiting and less constipation.
It should not be used concomi-tantly with monoamine oxidase inhibitors due to
potentially elevated levels of norepinephrine.
Propoxyphene with and without acetamino-phen (Darvocet and Darvon) was
withdrawn from the U.S. market in 2010 due to the risk of cardiac toxicity.
A. Parenteral Opioid Administration
Intravenous, intraspinal (epidural or
intrathecal), or transdermal routes of opioid administration must be utilized
when the patient fails to adequately respond to, or is unable to tolerate, oral
regimens. However, when the patient’s pain increases significantly, or changes
markedly in quality, it is equally impor-tant to reevaluate the patient for
adequacy of pain diagnosis and for the potential of disease progres-sion. In
patients with cancer, adjunctive treatments such as surgery, radiation,
chemotherapy, hormonal therapy, and neurolysis may be helpful. Intramuscu-lar
opioid administration is rarely optimal because of variability in systemic
absorption and resultant delay and variation in clinical effect.
B. Intravenous Opioid Therapy
Parenteral opioid therapy is usually best accom-plished by intermittent or continuous intravenous infusion, or both, but can also be given subcuta-neously. Modern portable infusion devices have patient-controlled analgesia (PCA) capability, allow-ing the patient to self-treat for breakthrough pain.
C. Spinal Opioid Therapy
The use of intraspinal opioids is an
excellent alter-native for patients obtaining poor relief with other analgesic
techniques or who experience unaccept-able side effects. Epidural and
intrathecal opioids offer pain relief with substantially lower total doses of
opioid and fewer side effects. Continuous infusion techniques reduce drug
requirements (compared with intermittent boluses), minimize side effects, and
decrease the likelihood of catheter occlusion. Myoclonic activity may be
occasionally observed with intrathecal morphine or hydromorphone.
Epidural or intrathecal catheters can be
placed percutaneously or implanted to provide long-term effective pain relief.
Epidural catheters can be attached to lightweight external pumps that can be
worn by ambulatory patients. A temporary catheter must be inserted first to
assess the poten-tial efficacy of the technique. Correct placement of the
permanent catheter should be confirmed using fluoroscopy with contrast dye.
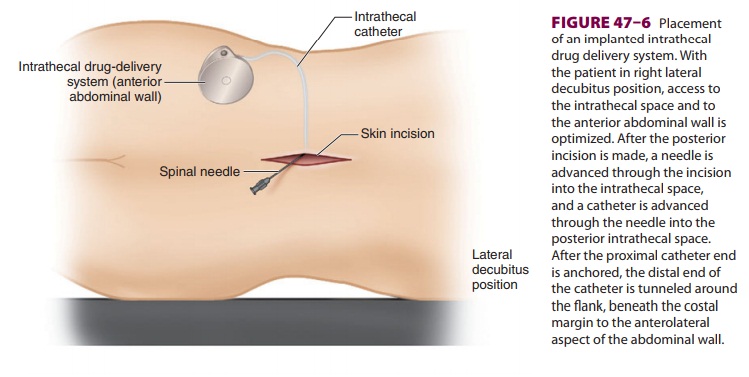
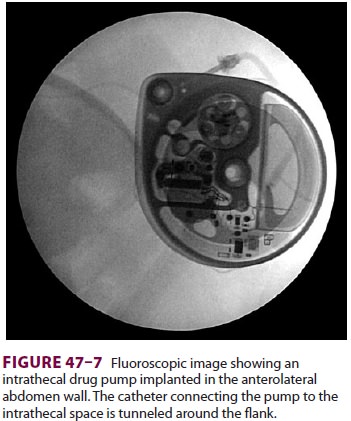
Completely implantable intrathecal catheters with externally programmable pumps
can also be used for con-tinuous infusion (Figure 47–6). The reservoir of the implanted pump (Figure 47–7) is periodicallyrefilled percutaneously. Implantable systems are most
appropriate for patients with a life expectancy of several months or longer,
whereas tunneled epi-dural catheters are appropriate for patients expected to
live only weeks. Formation of an inflammatory
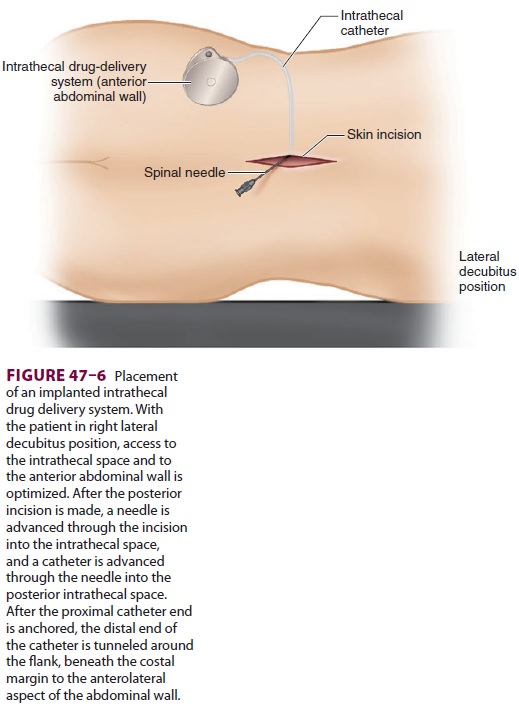
mass (granuloma) at the tip of the intrathecal cath-eter can occur and
may reduce efficacy.
The most frequently encountered problem
asso-ciated with intrathecal opioids is tolerance. Gener-ally a slow
phenomenon, tolerance may develop rapidly in some patients. In such instances,
adjuvant therapy must be used, including the intermittent use of local
anesthetics or a mixture of opioids with local anesthetics (bupivacaine or
ropivacaine 2–24 mg/d), clonidine (2–4 mcg/kg/h or 48–800 mcg/d,
respec-tively), or the GABA agonist baclofen. Clonidine is particularly useful
for neuropathic pain. In high doses, it is more likely to be associated with
hypo-tension and bradycardia.
Complications of spinal opioid therapy
include local skin infection, epidural abscess, meningitis, and death or
permanent injury from pump programming or drug dilution errors. Superficial
infections can be reduced by the use of a silver-impregnated cuff close to the
exit site. Other complications of spinal opi-oid therapy include epidural
hematoma, which may become clinically apparent either immediately fol-lowing
catheter placement or several days later, and respiratory depression.
Respiratory depression sec-ondary to spinal opioid overdose can be treated by
decreasing the pump infusion rate to its lowest set-ting and initiating a
naloxone intravenous infusion.
D. Transdermal Fentanyl
Transdermal fentanyl (Duragesic patch) is an alter-native to
sustained-release oral morphine and oxycodone preparations, particularly when
oral medication is not possible. The currently available patches are
constructed as a drug reservoir that is separated from the skin by a
microporous rate-limiting membrane and an adhesive polymer. A very large
quantity of fentanyl (10 mg) provides a large force for transdermal diffusion.
Transder-mal fentanyl patches are available in 25, 50, 75, and 100 mcg/h sizes
that provide drug for 2–3 days. The largest patch is equivalent to 60 mg/d of intravenous
morphine. The major obstacle to fentanyl absorp-tion through the skin is the
stratum corneum. Because the dermis acts as a secondary reservoir, fentanyl
absorption continues for several hours after the patch is removed. The
transdermal route avoids hepatic first-pass metabolism.
Major disadvantages of the transdermal route
are its slow rate of drug delivery onset and the inabil-ity to rapidly change
dosage in response to changing opioid requirements. Blood fentanyl levels rise
and reach a plateau in 12–40 h, providing average con-centrations of 1, 1.5,
and 2 ng/mL for the 50, 75, and 100 mcg/h patches, respectively. Large
inter-patient variability results in actual delivery rates ranging from 50 to
200 mcg/h. This formulation is popu-larly “diverted” for nonmedical uses and
has been the cause of numerous deaths from “recreational” pharmacology.
Botulinum Toxin (Botox)
OnabotulinumtoxinA (Botox) injection has been increasingly utilized in
the treatment of pain syn-dromes. Studies support its use in the treatment of
conditions associated with involuntary muscle con-traction (eg, focal dystonia
and spasticity), and it is approved by the FDA for prophylactic treatment of
chronic migraine headache. This toxin blocks ace-tylcholine released at the synapse
in motor nerve endings but not sensory nerve fibers. Proposed mechanisms of
analgesia include improved local blood flow, relief of muscle spasms, and
release of muscular compression of nerve fibers
Related Topics