Chapter: Biology of Disease: Disorders of the Endocrine System
Growth Hormone Disorders
GROWTH HORMONE DISORDERS
Growth hormone (GH) or somatotrophin promotes linear growth and the maintenance of tissues by stimulating the uptake of amino acids by cells, protein synthesis, increasing blood glucose concentration and fat metabolism and promoting epiphyseal bone growth. These effects are mediated by locally acting effectors called somatomedins that are synthesized by many tissues but particularly liver. Somatomedins stimulate cell proliferation and/or differentiation. They include insulin-like growth factors-I and II (IGF-I and IGF-II). Insulin-like growth factor-I is the most significant physiologically and, indeed, its concentration correlates with that of GH.
Growth hormone is a polypeptide 191 amino acid residues long (Figure 7.14(A)). Approximately 70% of plasma GH is bound to growth hormone bindingprotein. Growth hormone is synthesized in the anterior pituitary gland in
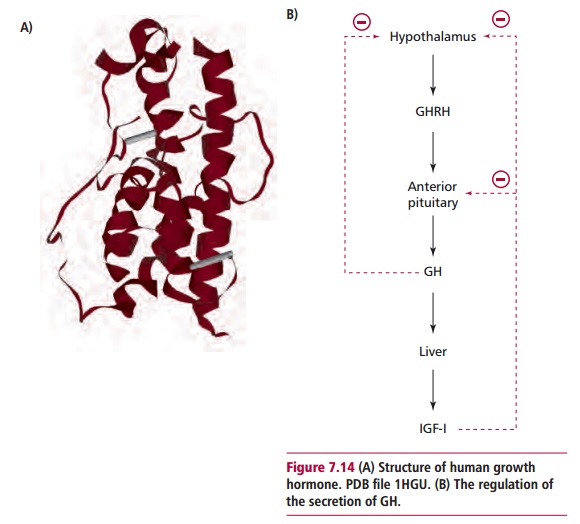
an inactive form called preprogrowth hormone that is hydrolyzed in several enzyme-catalyzed reactions to give active GH prior to its secretion. A number of factors, such as sleep, amino acids, exercise and stress stimulate GHRH release from the hypothalamus and that, in turn, stimulates GH secretion. Hyperglycemia stimulates the secretion of somatostatin from the hypo-thalamus and this inhibits the secretion of GH. Increasing concentrations of serum GH and IGF-I exert a negative feedback effect that prevents further release of GH (Figure 7.14 (B)). The rate of adult secretion varies but is generally about 1.4 mg daily and occurs in pulses with the largest amounts being released during sleep.
The clinical features resulting from an excess or deficiency of GH depend on the age of the person. A deficiency during childhood leads to a stunted growth called dwarfism (Figure 7.15 (A)) and therefore requires early detection. However, GH deficiency is a rare cause of dwarfism and other causes, for example thyroid deficiency or inadequate nutrition, need to be excluded first. The commonest causes of GH deficiency are nonendocrine tumors that affect the pituitary gland or hypothalamus. Growth hormone deficiency may also be a consequence of generalized pituitary disease or a congenital defect leading to a deficient production of GHRH. Whatever the cause, the major clinical feature is stunted growth, well below that expected for a child of comparable age, with a short height, immature face and skeleton as revealed by radiological investigations. Clinical signs of other anterior pituitary hormone deficiencies may be evident.
The most common cause of excessive GH release is a GH secreting pituitary tumor or adenoma . Although these are benign, they are not subject to normal control and continually release large amounts of GH. The causes of these tumors are unknown but a genetic basis has been suggested. Ectopic GH secretion is extremely rare but has been reported in patients with bronchial carcinoma.
Excessive GH release causes gigantism in children and acromegaly in adults. Children with GH excess grow as much as 6 inches per year to abnormal heights, often in excess of 8 feet. Muscle weakness is seen in longstanding cases. Acromegaly has an insidious onset and may take years for its clinical features, enlargement of bones of hands, feet and face, thickening of soft tissues causing coarse facial features, enlarged tongue and lips, prognathism or protruding jaw, increased sweating and enlargement of internal organs, such as liver, spleen and heart, to become apparent. Additionally, acromegalics suffer from paresthesia of the hands and feet due to entrapment of nerves by thickened bone and subcutaneous tissue, headaches/vision disorders due to the growing pituitary tumor and sensory nerve entrapment, impaired glucose tolerance or diabetes mellitus and increased incidences of coronary heart disease and stroke . Individuals affected by excessive GH secretion throughout life show features of both gigantism and acromegaly (Figure 7.15 (B)). The prognosis for both gigantism and acromegaly depends on how far the disorder has advanced. Gigantism is rarely life threatening and prognosis is usually good. However, an individual with advanced acromegaly will develop serious complications, such as coronary heart disease, cerebrovascular disease and diabetes mellitus.

DIAGNOSIS AND TREATMENT OF GROWTH HORMONE DISORDERS
A variety of tests are used to assess GH deficiency. Random measurements of serum GH are of limited value due to fluctuations in plasma GH levels in normal individuals. Urinary GH excretion is low in deficiency but obtaining an accurately timed collection of urine is difficult. Most tests rely on demonstrating that the hormone does not increase in concentration following a stimulus. Growth hormone increases after exercise and this has been used as a preliminary screening test. In the exercise test, the patient is subjected to hard physical exercise until they have a pulse rate greater than 150 beats per min . Blood is collected at 0, 2 and 20 min after stopping exercise. In normal individuals, the plasma concentration of GH increases by 20 mU dm–3 above the initial value. Growth hormone release increases during sleep, hence high values in a nocturnal sample may exclude deficiency. Blood samples are collected using a venous catheter at 30 min intervals for 3 to 4 h after the onset of sleep. A peak of at least 10 mU dm–3 occurs in normal individuals but not in patients with GH deficiency. Clonidine is a potent stimulator of GH secretion and is used in a definitive test for GH deficiency. Growth hormone from genetically engineered sources is used in treatment but must be continued until longitudinal growth is completed. In cases where deficiency is due to low levels of GHRH, analogs of this peptide, for example hexarelin, have been used.
A diagnosis of excess GH is made on clinical grounds supported by biochemical and radiological investigations. Photographs taken of the patient when younger are particularly useful when making a diagnosis. Basal serum concentrations of GH are increased in GH excess but, because release is influenced by so many factors, the result of a single sample is unreliable.
The concentration of IGF-I in serum is raised in patients with acromegaly and is of diagnostic significance. The oral glucose tolerance test (OGTT) is used to confirm a diagnosis of acromegaly and is similar to that used for diagnosis of diabetes mellitus except that levels of plasma GH are also determined. In a healthy individual, the glucose load suppresses GH release to below 2 mU dm–3 by stimulating the release of somatostatin. In patients with acromegaly, this suppression is not seen.
The management of GH excess cannot reverse any clinical changes that have taken place prior to treatment. However, management is important because it improves survival and reduces deaths due to heart disease or stroke. Growth hormone secreting tumors are treated in one of three ways, the clinical decision depending on severity of disease, the age of the patient and the response to treatment. Surgical removal of the pituitary tumor may be attempted depending on its size and location with steps taken to minimize damage to the anterior pituitary. If GH levels do not normalize, other treatments must be considered. Radiotherapy is usually performed over a 4–6 week period by external irradiation using a cobalt source. Its effects are slow and, in some cases, hyposecretion of other pituitary hormones may occur. Drugs, such as bromocriptine, suppress GH release in acromegalics and are often used in patients too old to undergo surgery or radiotherapy. Somatostatin analogs, for example octeotride, have also been used to inhibit GH release. Some patients require all three modes of treatment but are not always successfully treated. To detect recurrence, serum IGF-I needs to be monitored at regular intervals.
Related Topics