Chapter: Biology of Disease: Disorders of the Endocrine System
Congenital Adrenal Hyperplasia: Diagnosis and treatment - Disorders of the Adrenal Cortex
CONGENITAL ADRENAL HYPERPLASIA
Congenital adrenal hyperplasia (CAH) is an autosomal recessive condition characterized by an abnormal biosynthesis of steroid hormones in the adrenal glands. The clinical features depend on whether cortisol and/ or aldosterone or androgens are involved. The commonest cause of CAH is a complete or partial deficiency of 21-hydroxylase activity (Figure 7.32) that accounts for 95% of all cases, and occurs with an incidence of 1 in 12 000 newborn babies in the UK. A deficiency of 21-hydroxylase activity blocks the synthesis of cortisol and, as a consequence, negative feedback to the anterior pituitary is diminished. The anterior pituitary increases its secretion of ACTH, which causes hyperplasia of the adrenal glands. The substrate, 17 α -hydroxyprogesterone accumulates and stimulates the production of adrenal androgens. A deficiency in 21-hydroxylase activity is complete in approximately one-third of CAH patients. Less common forms of CAH are characterized by deficiencies of other enzymes in the synthetic pathway.
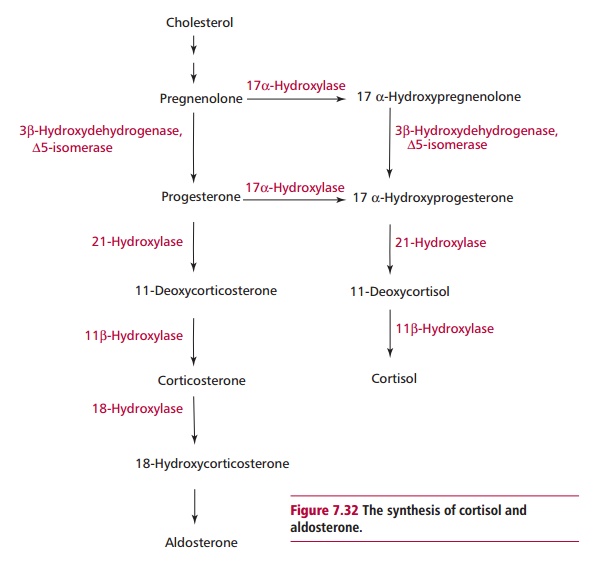
Females affected with CAH are born with ambiguous genitalia, although in cases of partial enzyme deficiency this may not be apparent until early adulthood where the female presents with hirsutism, amenorrhea and infertility. Males with CAH present with premature development of the male secondary sexual characteristics called pseudoprecocious puberty. In individuals with a complete deficiency of 21-hydroxylase activity, aldosterone production is also inhibited and these individuals usually present shortly after birth with a life-threatening condition characterized by excessive salt and water loss.
Diagnosis and treatment of congenital adrenal hyperplasia
Diagnosis of CAH due to 21-hydroxylase deficiency is made by detecting increased concentrations of 17 α -hydroxyprogesterone in the baby’s blood at least two days following birth. Maternal 17 α -hydroxyprogesterone may still be present at two days postparturition, hence the need for the delay.
The affected individuals are treated with cortisol and, if necessary, aldosterone. The treatment should reduce ACTH secretion and therefore excessive androgen production. The treatment requires monitoring by regular measurements of plasma 17 α -hydroxyprogesterone.
Related Topics