Chapter: Biotechnology Applying the Genetic Revolution: RNA-Based Technologies
Antisense Oligonucleotides
ANTISENSE OLIGONUCLEOTIDES
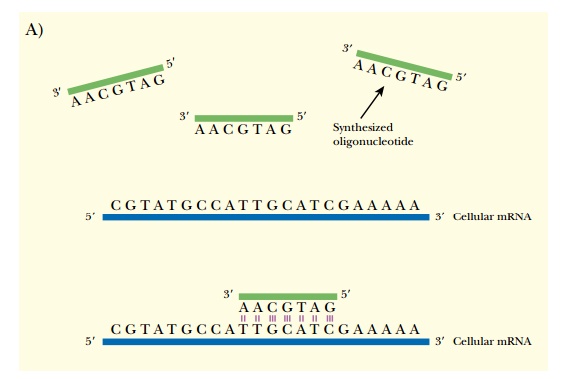
In the laboratory, antisense RNA can be made by two different methods (Fig. 5.3). The easiest method is to chemically synthesize oligonucleotides that are complementary to the target gene. The oligonucleotides are then injected or transformed into the target cell (see later discussion). Alternatively, the gene of interest can be cloned in the opposite orientation, so that transcription gives antisense RNA. The vector carrying the anti-gene is then transformed into the target organism.
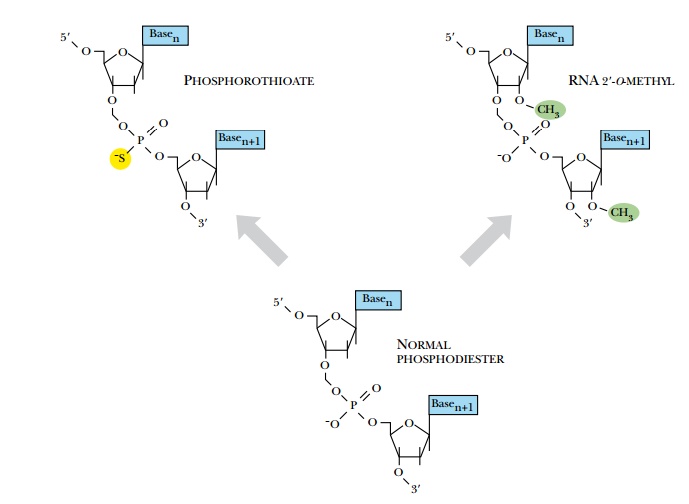
Chemically synthesized antisense oligonucleotides are traditionally made of DNA rather than RNA for two reasons. DNA is more stable in the laboratory, and DNA synthesis is an established and automated procedure. In the cell, DNA oligonucleotides are still very susceptible to degradation by endonucleases; therefore, various chemical modifications are added to increase intracellular stability. The most common modification is to replace one of the nonbridging oxygens in the phosphate groups with sulfur (Fig. 5.4) to make a phosphorothioate oligonucleotide. This makes the phosphorus a chiral center; one diastereomer is resistant to nuclease degradation but the other is still sensitive, leaving about half of the antisense molecules functional inside the cell. This modification does not affect the solubility of the oligonucleotides or their susceptibility to RNase H degradation. These types of antisense oligonucleotides have been developed to inhibit cancers such as melanoma and some lung cancers. The most common side effect with phosphorothioate oligonucleotides is nonspecific interactions, especially with proteins that interact with sulfur-containing molecules.
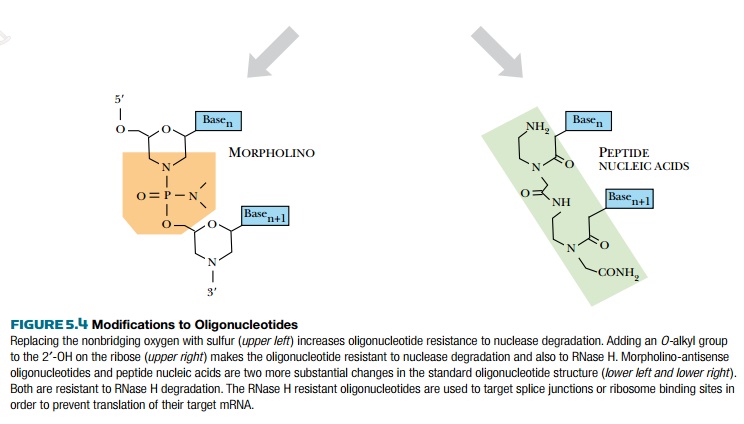
Two other modifications have fewer nonspecific interactions than phosphorothioate oligonucleotides. Adding an O-alkyl group to the 2′-OH of the ribose makes the oligonucleotide resistant to DNase and RNase H degradation (see Fig. 5.4). Inserting an amine into the ribose ring, thus changing the five-carbon ribose into a morpholino ring, creates morpholino-antisense oligonucleotides (see Fig. 5.4). In addition to the morpholino ring, a second amine replaces the nonbridging oxygen to create a phosphorodiamidate linkage. This amine neutralizes the charged phosphodiester of typical oligonucleotides. The loss of charge affects their uptake into cells, but alternative methods have been developed to get these antisense molecules into the cells (see later discussion). Both types of modified oligonucleotides are resistant to RNase H. Therefore, they do not promote degradation of an mRNA:DNA hybrid target. Consequently, their use is restricted to blocking splicing sites in the pre-mRNA transcript or to block ribosome binding sites.

The most different modified oligonucleotides are peptide nucleic acids (PNAs), which have the standard nucleic acid bases attached to a polypeptide backbone (normally found in proteins) rather than a sugar-phosphate backbone (see Fig. 5.4). The polypeptide backbone has been modified so that the RNA bases are spaced at the same distance as the typical oligonucleotide. The spacing is critical to function because the bases of a PNA must match the bases in the target RNA. This molecule is also uncharged and works through non-RNase H dependent mechanisms as with morpholino-antisense oligonucleotides. Antisense PNA has been developed to inhibit translation of the HIV viral transcript, gag-pol, and to block translation of two cancer genes, Ha-ras and bcl-2.
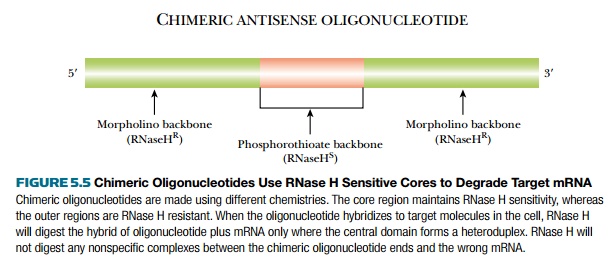
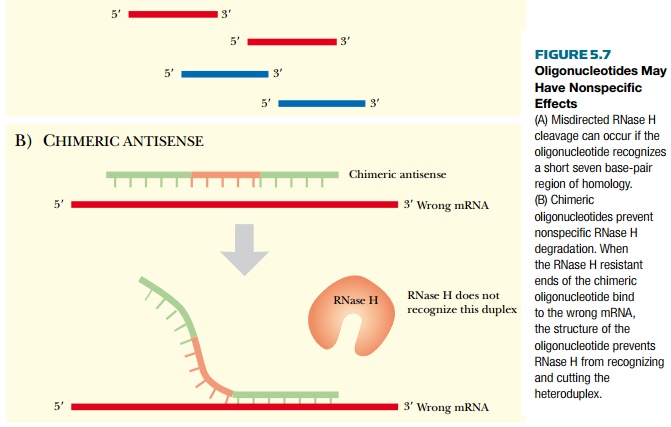
As noted earlier, antisense oligonucleotides that are resistant to RNase H must be made to target splice sites and/or ribosome binding sites in order to block the target mRNA. In some cases these sequences are not well characterized in the target mRNA, so modified antisense oligonucleotides become useless. Making mixed or chimeric antisense oligonucleotides can restore the targeting of RNase H to the mRNA and allow the researcher to use the modified structures to prevent degradation (see later discussion). In these chimeric antisense oligonucleotides, the core has a short (∼7) base-pair span of phosphorothioate linkages, which are RNase H sensitive, flanked on each side by sequences consisting of one of the RNase H–resistant modifications (Fig. 5.5). The flanking regions contain 2′-O-methyl groups, morpholino structures, or even PNA. These chimeric molecules can target any accessible regions of the mRNA, not just splice sites or ribosome binding sites.
Antisense Oligonucleotides May Alter Splicing
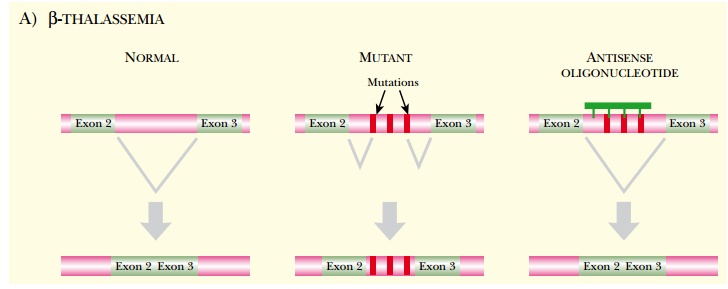
Alternative splicing is widely used to control gene expression in eukaryotes. Some diseases are caused by aberrant patterns of splicing. For example, beta-thalassemia is a blood disorder where red blood cells cannot carry enough oxygen because of defective hemoglobin. Some cases of the disorder arise from aberrant splicing of the beta-globin pre-mRNA. Antisense morpholino-oligonucleotides have been designed that target the mutant splice site between exons two and three in the beta-globin gene. The antisense oligonucleotide corrects the splicing pattern, and restores the correct protein in red blood cells taken from patients with beta-thalassemia (Fig. 5.6A).
The Bcl-x gene in humans produces two different proteins because of alternate splicing. Bcl-xL, the longer protein, includes a segment of coding material between exons 1 and 2. Bcl-xS, the shorter protein, lacks this segment. Bcl-xL protein promotes cell growth by blocking apoptosis, whereas Bcl-xS opposes this and causes cells to die via apoptosis. In some cancers, the long form is overexpressed. Thus, devising a method to inhibit Bcl-xL expression and enhance Bcl-xS expression could induce these cancer cells to undergo apoptosis and die. Antisense 2′-O-methyl-oligonucleotides to the splice site in the pre-mRNA prevent the longer form from being made. The Bcl-xS protein then counteracts the cancerous growth by promoting apoptosis (see Fig. 5.6B). Those cancer cells that were “fixed” by antisense therapy also became more sensitive to chemotherapeutic agents because of the restoration of apoptosis.
Problems Associated with Antisense Oligonucleotides
There are two major problems associated with antisense technology. The most important is the design of the antisense oligonucleotide itself. Although mRNA is usually depicted as a linear molecule, mRNA can fold back on itself, forming normal Watson-Crick base pairs between different regions. In addition, some regions form loops and turns, creating a
three-dimensional structure. Because some target regions are annealed to other regions via base pairing, and other target regions may be hidden deep within the three-dimensional structure, finding a region of the target mRNA accessible to a laboratory-designed antisense oligonucleotide can be difficult.
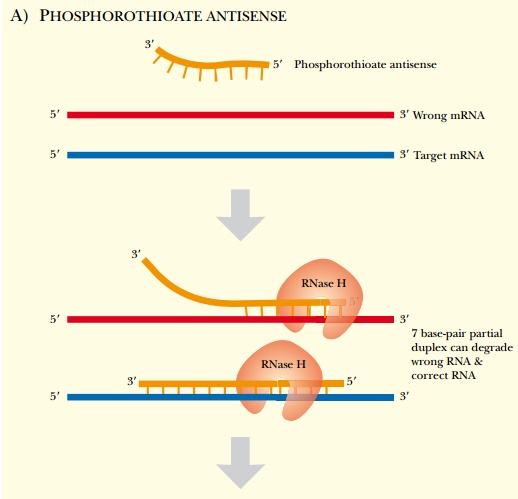
The second major challenge with antisense technology is nonspecific interactions with nontarget molecules. As mentioned earlier, phosphorothioate oligonucleotides have a propensity to bind cellular proteins that interact with sulfur-containing compounds, essentially pulling the oligonucleotides away from the target mRNA. In addition, small stretches of an oligonucleotide may base-pair with nonspecific mRNA sequences. Normally, this would not pose any problem, because the heteroduplex region is short and unstable. However, if the oligonucleotides are sensitive to RNase H, this endonuclease may degrade the wrong mRNA. The extent of homology between RNA and DNA only needs to be about seven base pairs for RNase H recognition, so when a short unstable heteroduplex forms, it can be sufficient to activate RNase H. In order for the antisense oligonucleotide to specifically target only one mRNA, the region of homology needs to be about 20 bases in length to be specific. Using chimeric antisense oligonucleotides is one method to overcome this problem, because RNase H can cleave only the core region of the chimera (Fig. 5.7). Even if the ends of the chimeric antisense oligonucleotide find a seven-base-pair region of homology with a nontarget mRNA, RNase H cannot cleave these nonspecific targets.
Related Topics