Chapter: Ophthalmology: Retina
Retinal Dystrophies: Retinitis Pigmentosa
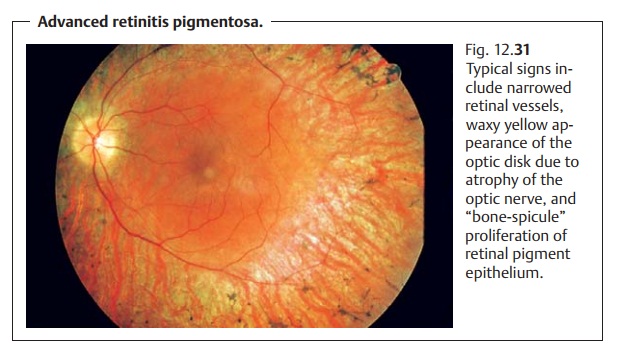
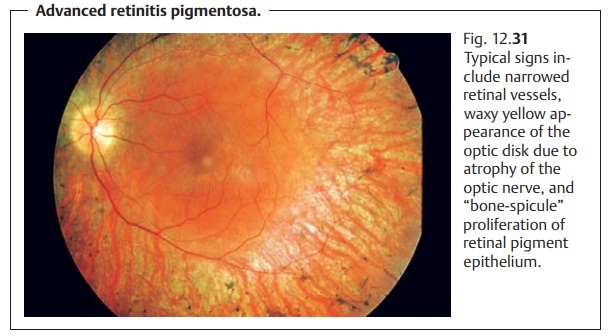
Retinitis Pigmentosa
Definition
This term is used to refer to a heterogeneous group of retinal disorders that lead to progressive loss of visual acuity, visual field defects, and night blind-ness. The name retinitis pigmentosa comes from the pigment deposits that characterize these disorders. In their classic form (see findings and diagnostic considerations) of such disorders, these deposits progress from the periphery to the center of the retina.
Epidemiology:
The worldwide incidence of retinitis pigmentosa is estimatedat between one per 35000 and one per 70000 persons. The estimated inci-dence of mutated alleles is one per 80 persons.
Forms of retinitis pigmentosa:
1. Rod-cone dystrophy (classic retinitis pigmentosa, by far the most frequent form).
2. Cone-rod dystrophy (inverse retinitis pigmentosa).
3. Sectoral retinitis pigmentosa
4. .Retinitis pigmentosa sine pigmento (form without pigment).
5. Unilateral retinitis pigmentosa.
6. Leber’s amaurosis (form occurring in early childhood).
7. Retinopathy punctata albescens (punctate retinitis).
8. In combination with other disorders in syndromes and metabolic dis-orders such as mucopolysaccharidoses, Fanconi’s syndrome, mucolipido-sis IV, peroxisomal disorders, Cockayne’s syndrome, mitochondrial
myopathies, Usher’s syndrome, neuronal and ceroid lipofuscinoses, renal tubular defect syndromes, etc.
Retinitis pigmentosa occurs almost exclusively as rod-cone dystrophy. There-fore, the other extremely rare forms are not discussed here except for the inverse form of classic retinitis pigmentosa, which is presented for purposes of comparison.
Inheritance:
Individual genetic forms may be identified from among the het-erogeneous group of disorders comprising retinitis pigmentosa. This group of disorders can involve various genotypes as well as variable phenotypic expression or different stages of a disorder with one specific genotype. There are over 15 purely ocular forms of retinitis pigmentosa. The most common form of inheritance is autosomal recessive (60%), followed by autosomal dominant (up to 25%), and X-linked (15%). Rhodopsin gene mutations (chro-mosome 3) and “retinal degeneration slow” (RTS) gene mutations (chromo-some 6) have also been described.
Symptoms:
Initial symptoms of retinitis pigmentosa include glare, nightblindness, progressive visual field defects, loss of visual acuity, and color vision defects. The age of manifestation depends on the type of inheritance.
Findings and diagnostic considerations:
The diagnosis is made by ophthal-moscopy on the basis of a classic picture.
Rod-cone dystrophy (primarily the rods are affected first). “Bone-spicule”proliferation of retinal pigment epithelium is observed in the middle peripheryof the retina. This will gradually spread toward the center and farther periph-erally (Fig. 12.31). Early deficits include color vision defects and disturbed contrast perception. Atrophy of the optic nerve, discernible as a waxy yellow appearance of the optic disk, will occur in the advanced stages. The arterieswill appear narrowed, and the fundus reflex will be extremely muted. Thepatient will typically have a “gun-barrel” visual fieldwith good visual acuity for a surprisingly long time but with progressive loss of the peripheral visual field.
Cone-rod dystrophy (primarily the cones are affected first). Here, there isearly loss of visual acuity with gradual progressive loss of visual field. In both forms of retinitis pigmentosa, the diagnosis is confirmed by electroretinogra-phy. Light response in the electroretinogram will be sharply reduced or absent early in the clinical course of the disease.
Differential diagnosis:
Differential diagnosis should consider changes col-lectively referred to as pseudoretinitis pigmentosa because they simulate the clinical picture of retinitis pigmentosa. The most common causes that should be excluded in this context are:
âť– Posttraumatic changes.
❖ Postinflammatory or postinfectious changes. These may include degener-ative retinal pigment epithelial disease secondary to rubella with “salt and pepper” fundus of punctate areas of atrophy and proliferation of retinal pigment epithelium. Other causes include syphilis, which may present with placoid lesions of pigment epithelial atrophy and proliferations.
âť– Tumors.
âť– Medications, such as chloroquine, Myambutol (ethambutol), and thiori-dazine.
Treatment:
The causes of the disorder cannot be treated. Edge-filtered eye-glasses (eyeglasses with orange or blue colored lenses that filter out certain wavelengths) and magnifying near vision aids can help make better use of the patient’s remaining vision.
Prophylaxis:
No prophylaxis is possible.
Clinical course and prognosis:
Retinitis pigmentosa is chronically progress-ive. The clinical course depends on the specific form of the disorder; severe forms lead to blindness.
Related Topics