Chapter: Modern Analytical Chemistry: Gravimetric Methods of Analysis
Theory and Practice of Precipitation Gravimetry: Occlusions
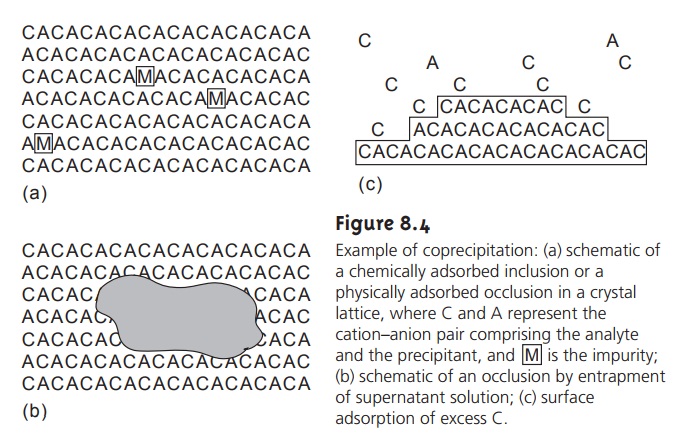
Occlusions
Which are a second
type of coprecipitated impurity, occur when physically adsorbed interfering ions
become trapped within
the growing precipitate.
Occlusions form in two ways.
The most common
mechanism occurs when physically
adsorbed ions are
surrounded by additional precipitate before they
can be desorbed or displaced (see Figure
8.4a). In this
case the precipitate’s mass is always
greater than expected. Occlusions also form when rapid precipitation traps a pocket of solution within the growing precipitate (Figure 8.4b). Since
the trapped solution
contains dis- solved solids,
the precipitate’s mass normally increases. The mass of the precipitate may be less than expected, however,
if the occluded material consists
primarily of the analyte in a lower-molecular-weight form
from that of the precipitate.
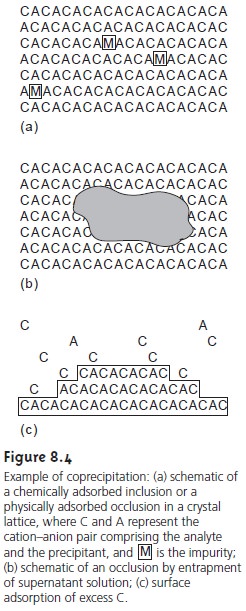
Occlusions are minimized by maintaining the precipitate in
equilibrium with its supernatant solution
for an extended time. This process is called digestion and may be carried out
at room temperature or at an elevated temperature. During di-
gestion, the dynamic nature of the solubility–precipitation equilibrium, in which
the precipitate dissolves and re-forms, ensures
that occluded material
is eventually exposed to the supernatant solution. Since the rate of dissolution and reprecipita-
tion are slow, the chance
of forming new occlusions is minimal.
After precipitation is complete the surface continues to attract ions from solu- tion (Figure 8.4c). These surface adsorbates,
which may be chemically or physically
adsorbed, constitute a third type of coprecipitated impurity. Surface adsorption is minimized by decreasing the precipitate’s available
surface area. One benefit of di-
gestion is that it also increases the average size of precipitate particles. This is not
surprising since the
probability that a particle will
dissolve is inversely proportional to its size. During digestion
larger particles of precipitate increase
in size at the ex- pense
of smaller particles. One consequence of forming fewer
particles of larger
size is an overall
decrease in the precipitate’s surface
area. Surface adsorbates also may be removed
by washing the precipitate. Potential
solubility losses, however,
cannot be ignored.
Inclusions, occlusions, and surface adsorbates are called
coprecipitates because they represent soluble
species that are brought into solid form along with the de- sired precipitate. Another source
of impurities occurs
when other species
in solu- tion precipitate under the conditions of the analysis. Solution conditions necessary to minimize the solubility of a desired
precipitate may lead
to the formation of an additional precipitate that interferes in the analysis. For example, the precipitation
of nickel dimethylgloxime requires a pH that
is slightly basic.
Under these condi- tions, however, any Fe3+ that might
be present precipitates as Fe(OH)3. Finally, since most precipitants are not selective
toward a single analyte, there is always a
risk that the precipitant will react, sequentially, with more than one species.
The
formation of these
additional precipitates can usually be minimized by carefully controlling solution conditions. Interferents forming precipitates that are less soluble
than the analyte
may be precipitated and removed
by filtration, leaving the analyte behind in solution. Alternatively, either the analyte
or the interferent can be masked using
a suitable complexing agent, preventing its precipitation.
Both of the above-mentioned approaches are illustrated in Fresenius’s analyti- cal method for determining Ni and Co in ores
containing Pb2+, Cu2+,
and Fe3+ as potential interfering ions. The
ore is dissolved in a so- lution containing H2SO4, selectively precipitating Pb2+ as PbSO4. After filtering, the supernatant solution is treated with H2S. Because
the solution is strongly acidic,
however, only CuS precipitates. After
removing the CuS by filtration, the solution is made
basic with ammonia
until Fe(OH)3 precipitates. Cobalt and nickel,
which form soluble amine
complexes, remain in solution.
In some situations the rate at which a precipitate forms
can be used to separate an analyte from a potential interferent. For example, due to similarities in their chemistry, a gravimetric analysis for Ca2+ may be adversely affected by the presence of Mg2+. Precipitates of Ca(OH)2, however, form more rapidly
than precipitates of Mg(OH)2. If Ca(OH)2
is filtered before Mg(OH)2 begins to precipitate, then a
quantitative analysis for Ca2+ is
feasible.
Finally, in some cases it is easier
to isolate and weigh both the analyte
and the interferent. After recording its weight, the mixed precipitate is treated to convert at least
one of the two precipitates to a new chemical form. This new mixed precipitate is also isolated and weighed. For example, a mixture containing Ca2+ and Mg2+ can be analyzed for both cations by first isolating a mixed precipitate of CaCO3 and MgCO3. After
weighing, the mixed
precipitate is heated,
converting it to a mixture of CaO and MgO. Thus
Grams of mixed precipitate 1 = grams CaCO3 + grams
MgCO3
Grams of mixed precipitate 2 = grams CaO + grams MgO
Although these equations contain four unknowns (grams CaCO3,
grams MgCO3, grams CaO, and grams MgO), the stoichiometric relationships between CaCO3 and CaO
Moles CaCO3 = moles CaO
and between MgCO3 and MgO
Moles MgCO3 = moles MgO
provide enough additional information to determine the amounts of both Ca2+ and
Mg2+ in
the sample.*
Related Topics