Chapter: Modern Analytical Chemistry: Gravimetric Methods of Analysis
Theory and Practice of Precipitation Gravimetry: Controlling Particle Size
Controlling Particle Size
Following precipitation and digestion, the precipitate
must be separated from the supernatant solution
and freed of any remaining impu- rities, including residual solvent. These tasks
are accomplished by filtering, rinsing, and drying the precipitate. The size of the precipitate’s particles determines the
ease and success of filtration. Smaller,
colloidal particles are difficult to filter because they may readily pass through the pores of the filtering device. Large, crystalline particles, however,
are easily filtered.
By
carefully controlling the precipitation reaction
we can significantly increase
a precipitate’s average
particle size. Precipitation consists of two
distinct events: nu- cleation, or the initial
formation of smaller
stable particles of precipitate, and the
subsequent growth of these particles. Larger particles form when the rate of particle
growth exceeds the rate of nucleation.
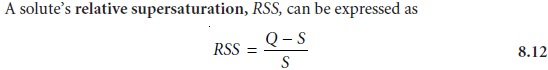
where Q is the
solute’s actual concentration, S is the solute’s expected concentra-
tion at equilibrium, and Q –
S
is a measure
of the solute’s supersaturation when
precipitation begins.
A large, positive
value of RSS indicates that a solution
is highly supersaturated. Such solutions are unstable and show high rates of nucle- ation, producing a precipitate consisting of numerous small particles. When RSS is small, precipitation is more likely to occur by particle growth than by nucleation.
Examining equation 8.12 shows that we can minimize RSS by
either decreasing the solute’s
concentration or increasing the precipitate’s solubility. A precipitate’s
solubility usually increases
at higher temperatures, and adjusting pH may affect a
precipitate’s solubility if it contains
an acidic or basic anion.
Temperature and pH, therefore, are useful ways to increase
the value of S. Conducting the precipitation in a dilute solution
of analyte, or adding the precipitant slowly
and with vigorous
stir- ring are ways
to decrease the
value of Q.
There are, however, practical limitations to minimizing RSS. Precipitates that are extremely insoluble, such as Fe(OH)3 and PbS, have such small
solubilities that a large
RSS cannot be avoided.
Such solutes inevitably form small particles. In addi- tion, conditions that yield a small RSS may
lead to a relatively stable
supersaturated solution that requires
a long time to fully precipitate. For example, almost a month is
required to form a visible
precipitate of BaSO4 under conditions in which the ini-
tial RSS is 5.
An increase in the time required to form a visible precipitate under conditions of low RSS is a consequence of both a slow rate of nucleation and a steady decrease
in RSS as the
precipitate forms. One
solution to the
latter problem is to chemically generate the precipitant in solution as the product
of a slow chemical reaction. This maintains the RSS at
an effectively constant level. The precipitate initially forms
under conditions of low RSS, leading to the nucleation of a limited
number of parti- cles. As additional precipitant is created, nucleation is eventually superseded by par- ticle growth.
This process is called homogeneous precipitation.
Two general methods
are used for
homogeneous precipitation. If the precipi- tate’s solubility is pH dependent, then the analyte
and precipitant can be mixed under conditions in which
precipitation does not occur. The pH is then raised
or lowered as needed by chemically generating OH– or
H3O+. For example,
the hydrol- ysis of urea can
be used as a source
of OH–.
CO(NH2)2(aq)+ H2O(l) < == == > CO2(g) + 2NH3(aq)
NH3(aq)+ H2O(l) < == == > NH4+(aq)+ OH–(aq)
The hydrolysis of urea is strongly temperature-dependent, with
the rate being
negli- gible at room temperature. The rate of hydrolysis, and thus the rate of precipitate
formation, can be controlled by adjusting the solution’s temperature. Precipitates of
BaCrO4, for example, have been produced in this manner.
In the second
method of homogeneous precipitation, the precipitant itself is generated by a chemical reaction. For example, Ba2+ can be homogeneously precipi- tated as BaSO4 by hydrolyzing sulphamic acid to produce
SO42–.
NH2SO3H(aq)+ 2H2O(l) < == == > NH4+(aq)+ H3O+(aq)+ SO42–(aq)
Homogeneous precipitation affords
the dual advantages of producing large particles of precipitate that are
relatively free from impurities. These advantages, however, may be offset
by increasing the time needed
to produce the precipitate,
and a tendency for the precipitate to deposit as a thin film on the container’s walls. The latter problem
is particularly severe for hydroxide
precipitates generated using urea.
An additional method
for increasing particle
size deserves mention.
When a precipitate’s particles are electrically neutral,
they tend to coagulate into larger par- ticles. Surface adsorption of excess lattice
ions, however, provides
the precipitate’s particles with a net positive or negative surface
charge. Electrostatic repulsion
be- tween the particles prevents them from
coagulating into larger
particles.
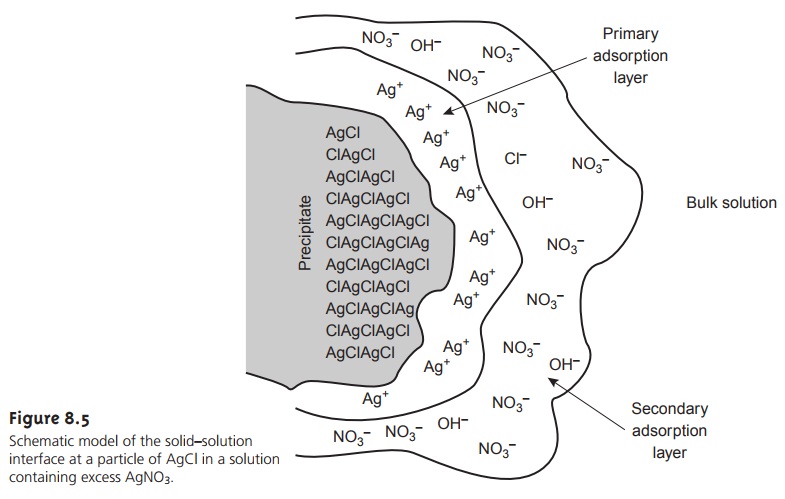
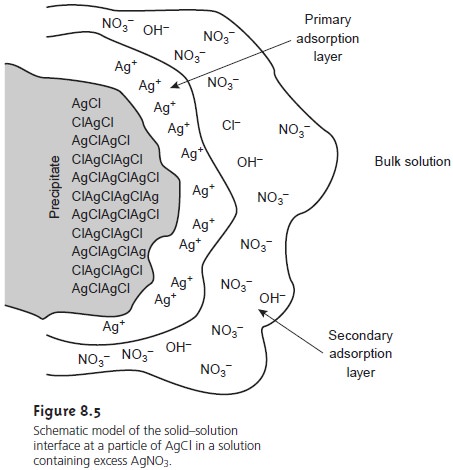
Consider, for instance,
the precipitation of AgCl from a solution
of AgNO3, using NaCl
as a precipitant. Early in the precipitation, when NaCl is the limiting reagent, excess Ag+ ions chemically adsorb to the AgCl
particles, forming a posi-
tively charged primary
adsorption layer (Figure
8.5). Anions in solution, in this case NO3– and OH–,
are attracted toward
the surface, forming
a negatively charged
sec- ondary adsorption layer that balances the surface’s positive
charge. The solution outside the secondary adsorption layer remains
electrically neutral. Coagulation cannot occur if the secondary adsorption layer is too thick because
the individual particles of AgCl are unable to approach one another closely
enough.
Coagulation can be induced in two ways: by increasing the concentration of the ions responsible for the secondary adsorption layer or by heating the solution. One way to induce coagulation is to add an inert electrolyte, which increases the concen- tration of ions in the secondary adsorption layer. With more ions available, the thickness of the secondary absorption layer decreases. Particles of precipitate may now approach one another more closely, allowing the precipitate to coagulate. The amount of electrolyte needed to cause spontaneous coagulation is called the critical coagulation concentration.
Heating the solution
and precipitate provides
a second way to induce
coagula- tion. As the temperature increases, the number of ions in the
primary adsorption layer decreases, lowering the precipitate’s surface charge. In addition, increasing the particle’s kinetic energy may be sufficient to overcome
the electrostatic repulsion preventing coagulation at lower temperatures.
Related Topics