Chapter: Biochemical Pharmacology : Drugs that act on sodium and potassium channels
Local anesthetics
Local anesthetics
Sodium channels are
responsible for the propagation of action potentials in nerve fibers. Local
anesthetics are blockers of sodium channels. They will thus intercept the
propagation of action potentials along nerve fibers and in this way, among
other things, prevent perception of pain.
We have seen before that drug
receptors may be (in fact, typically are) allosteric molecules. This also
applies to voltage-gated channels. With these, the force or energy required for
transition from the resting to the active state is normally provided not by
ligand binding but by an elec-trical field. We have seen as well that drugs may
interact differentially with the inactive state and the active state of a
receptor. With voltage-gated channels, we actually have three different
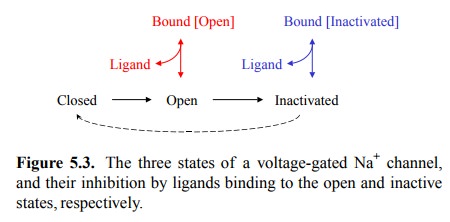
conformational states – they may be closed, open or inactivated. The functional
effects of local anes-thetics are related to their interactions with both the
open and the inactivated states (Figure 5.3). Interestingly, with the local
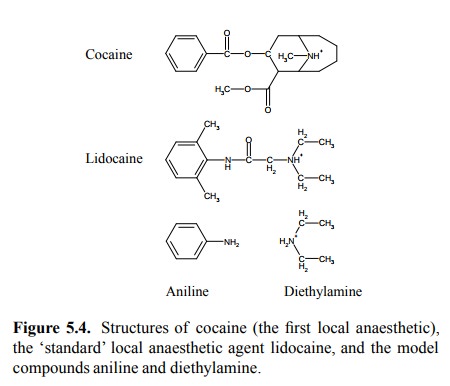
anesthetics of the lidocaine group, these two in-teractions can be assigned to
two different moieties of the drug molecule. These two moieties are represented
by ani-line and by diethylamine, respectively (Figure 5.4).
Binding of a drug to a
channel in its open state would be expected to obstruct the channel lumen to a
certain extent, depending on the location of the drug binding site relative to
the ion-conducting pathway (or channel lumen).
The effect of diethylamine
(DEA) on the conductance of a single NaV channel is depicted in
Figure 5.5a. In the control trace, the channel can be seen to oscillate between
two states of conductance, with currents of 0 and ~1 pA, respectively. A
conductance of 0 would be expected for the closed and the inactivated states,
respectively, whereas the conductance of 1 pA would represent the open state2.
This illustrates a very neat feature of the single channel recording techniques
– they let us observe the discrete and stochastic nature of conformational
changes of the proteins in a much more direct fashion than typically possible
with other allosteric proteins (e.g., enzymes or hormone recep-tors). In the
presence of DEA, open and closed state still alternate, but the conductance of
the open state is reduced by about 40%, indicating a partial blockade of the
channel.
Considering that DEA is a
cation itself, it seems likely that it acts by binding within and direct
obstruction of the chan-nel lumen (although an indirect mode of inhibition –
bind-ing outside the lumen, causing obstruction in an allosteric fashion –
cannot be ruled out).
A different pattern is
observed with aniline (Figure 5.5b). Here, the conductance of the open state is
unaltered; the ef-fect of the ligand instead consists in the occurrence of
ex-tended time intervals with zero conductance (note that the time scale
differs between Figure 5.5a and 5.5b), in line with the assumption that this
ligand binds to and stabilizes the inactivated state of the channel.
Interestingly, the chan-nel also shows brief closing intervals that resemble
those in the control trace. These might represent either direct rever-sions to
the closed state, or inactivation events that revert to the closed (yet not
inactivated) state before binding of aniline.

If the two moieties of
lidocaine exert distinguishable ef-fects on different states of the channel,
what is the spatial relationship between the two binding sites? Consider the
concentrations of aniline and diethylamine in the experients above. They are
very large – about 10 3-fold higher than the binding constant for
lidocaine (12 µM). The higher binding affinity of lidocaine then suggests that
the two low-er affinities of its components may combine – which could only work
if the two sites were adjacent to each other (Fig-ure 5.6).
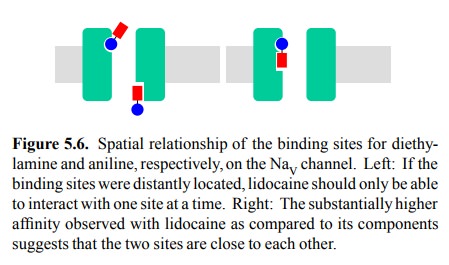
Figure 5.6. Spatial relationship of the binding sites for diethy-lamine and
aniline, respectively, on the NaV channel. Left: If the binding
sites were distantly located, lidocaine should only be able to interact with
one site at a time. Right: The substantially higher affinity observed with
lidocaine as compared to its components suggests that the two sites are close
to each other.
In Figure 5.3, we omitted the
possibility of lidocaine in-teracting with the resting (closed) sodium channel.
How do we know? Evidence of this is depicted in Figure 5.7c. It shows that the
fraction of channels ready to open in re-sponse to a sudden depolarization
depends on the level of the resting membrane potential before the depolariza-tion.
At levels below –90 mV, there isn't much of a dif-ference depending on whether
or not lidocaine is present – we would get a maximum response regardless. At
these very low membrane potentials (and sufficient time after the last opening
inactivation cycle), all channels will be in the closed stated and ready for
opening (cf. Figure 5.3). If li-docaine were able to arrest channels in the
closed state and prevent them from opening, we should see a reduced re-sponse
from any starting level of the membrane potential4.
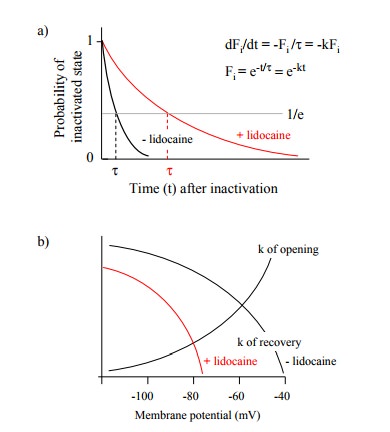

Figure 5.7. Inactivation of NaV channels by lidocaine. a: Firstorder kinetics of reactivation. b: Variation of the rate constants for channel opening and channel reactivation with the membrane potential. c: Fraction of NaV channels in the closed state (i.e., ready for opening in response to depolarization) as a function of the membrane potential. The diagrams are not drawn to scale.
How can we explain the shift
of the responsiveness of sodi-um channels to lower voltages? Figure 5.7a
illustrates the kinetic law of reactivation; it is a simple first order
process. If lidocaine is bound to the inactivated channel, the reacti-vation
rate constant is decreased. In addition, the reactiva-tion rate constant varies
with the membrane potential. So does the rate constant of channel opening;
however, the two vary in opposite ways, as illustrated in Figure 5.7b. At very
low membrane potentials, reactivation will always be fast relative to opening,
and the majority of channels will there-fore be in the closed state, ready to
open. As the potential increases, opening will get faster and reactivation
slower, and somewhere above -60 mV the balance will tilt, to that the lifetime
of the inactivated state now exceeds that of the closed state. This leads to a
depletion of the closed state. Since lidocaine reduces the rate of reactivation
at all mem-brane potentials (Figure 5.7b), the tipping of the balance will
occur at lower values, so that at the physiological rest-ing potential (-70 mV)
the closed state is already depleted, and no action potential will be
triggered.
It is interesting to note
that, even in the absence of lido-caine, the number of responsive channels
starts to drop be-tween the resting potential (–70 mV) and the firing level
(–55 mV). From this, we would expect a slow, partial depo-larization to render
the membrane refractory to excitation. This property of the NaV
channels may be important in a phenomenon called `depolarizing block' in
skeletal muscle cells. We will look into this in a later section.
Lidocaine is structurally
similar to cocaine, which was the first clinically useful local anaesthetic
(Figure 5.4). The stimulating effect of cocaine, however, is due to its effect
on a second, different receptor in the brain that indirectly amplifies the
effect of dopamine and norepinephrine (we will deal with this matter in a later
lecture). This effect is ac-tually observed at concentrations lower than those
required for the blocking of sodium channels. Yet, local application of cocaine
will result in very high concentrations that will easily exceed the threshold
for sodium channel blockade. Cocaine was first used in the eye, from where
uptake into the systemic circulation is quite insignificant. This also seems to
be its only remaining use in medicine. In oth-er applications such as spinal
and intravenous anaesthesia, which will lead to higher systemic drug
concentrations and therefore potentially more side effects, lidocaine and
simi-lar drugs have replaced cocaine.
Related Topics