Chapter: Biochemical Pharmacology : Drugs that act on sodium and potassium channels
Digitalis (foxglove) glycosides
Digitalis (foxglove)
glycosides
In other (mainly elderly)
patients, the main problem may consist not so much in under-perfusion of the
heart muscle but in a weak contractility. The heart becomes distended, further
reducing the effectiveness of contraction5. Here, we clearly want to
increase the contractility of the heart mus-cle. The most effective way to do
this is to raise the avail-ability of calcium in the cytosol. This is done with
digitalis (foxglove) glycosides. The mechanism of action of these is outlined
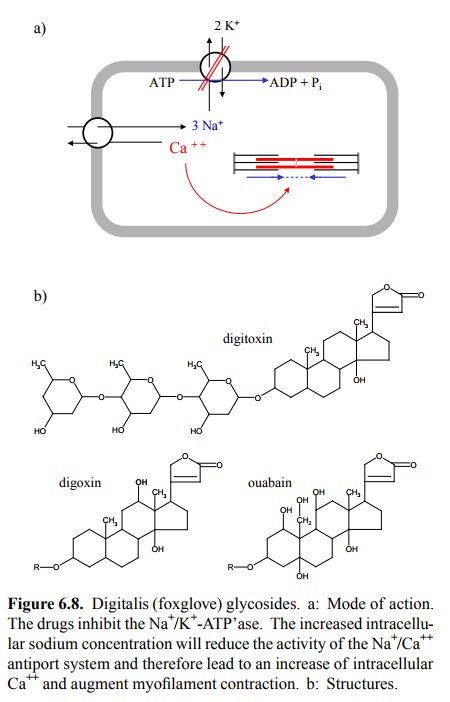
in Figure 6.8a. Digitalis glycosides bind to and block the Na+/K+-ATP'ase
in the cytoplasmic membrane. The backup of sodium in the cell reduces the
active export of Ca++ from the cytoplasm. Although the transfer of
cal-cium across the cytoplasmic membrane is small relative to the fluxes
occurring across the ER membrane, the ER stores will eventually fill up, and
the cytosolic Ca ++ level will be raised. This treatment is quite
effective and was readily recognized as such in an earlier era that did not
make use of advanced statistical methods to measure therapeutic re-sponses.
Nevertheless, the usage of digitalis is not undis-puted, particularly in North
America. Why? It is a matter of perspective. Patients usually benefit as long
as they live – but they do not live longer. Digitalis glycosides have, as you
will now have come to expect, effects on the pacemak er cells as well, and so
they do promote certain types of ar-rhythmias themselves.
If we consider the mode of
action of digitalis, what would we expect the therapeutic index of the drug to
be: Large or rather smaller? Complete receptor saturation will com-pletely
knock out the Na+/K+-ATP'ase and thereby termi-nate the life
of the target cell. Thus, it is quite obvious that we will have to walk a fine
line in determining the right dosage. Therefore, we will have to consider very
carefully the pharmacokinetic properties of the drugs, and the kidney and/or
liver functions of the patient.
Figure 6.8b compares the
three major digitalis glycosides that are (or have been) used clinically. The
structure of digitoxin is depicted completely, with both the steroid-like
`aglycone' moiety and the three residues of digitose, which is a hexose that
lacks two hydroxyl groups. These are rep-resented by `R' in the two other
structures and actually not required for activity – they can be replaced by,
e.g., a single acetyl residue. However, the lactone group – the five-mem-bered
ring at the top – is essential. Its hydrolysis (cleavage of the bold single
bond) or its reduction (of the bold dou-ble bond to a single bond) will abolish
activity. And here is a snag: Digitoxin, being highly protein-bound, is not
ef-ficiently eliminated in the kidneys but instead conjugat-ed in the liver and
largely secreted into the bile and intes-tine. There, a large fraction
undergoes cleavage of the (glu-curonide) conjugate and then reuptake, i.e.
enterohepatic cycling, so that this drug has a very long overall half-life.
However, during the intestinal passage, the lactone ring may be reduced by
bacterial enzymes, and the drug molecule thus be inactivated. Due to the
individual variations in the intestinal flora, this reduction may occur to
varying extents, which introduces an element of variability into the
effec-tiveness of this drug. Worse still, the analytical separation of the
reduced and the unreduced drug is not trivial and is not achieved by the
routine drug monitoring assays present-ly available.
Digoxin has one additional
hydroxyl group, which (some-what miraculously, it would seem) changes its
pharmacoki-netic parameters such that it is largely eliminated in the kid-neys.
This avoids the intricacies of entero-hepatic cycling; however, it renders the
rate of elimination dependent on kidney function, which tends to be more
variable than liver function. Also, intestinal uptake directly after oral
inges-tion tends to be lower and more variable. An even more polar derivative
is ouabain. This drug is rapidly eliminated in the kidneys. However, it is not
efficiently taken up after oral application and so can only be used
intravenously.
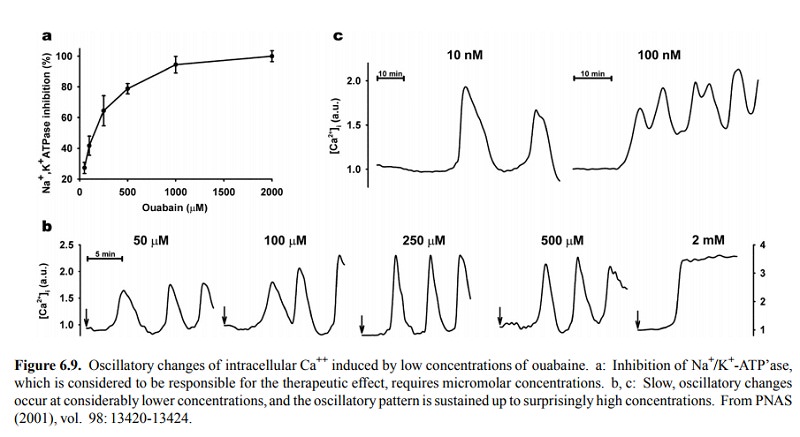
Recently, it has been found
that ouabain (once thought to occur in plants only) is, in fact, secreted by
the adrenal glands – at concentrations far lower than those necessary for
inhibition of the Na+/K+-ATPase. Nevertheless, there is experimental evidence
that even at the low physiological concentrations, ouabain may modulate
cellular function. This is illustrated in Figure 6.9. While half-maximal
inhibi-tion of Na+/K+-ATP'ase occurs at about 100-200 µM
(Fig-ure 6.9a), elevations in the cellular calcium level (measured using a
cell-permeant, calcium-binding fluorescent dye) are detected with nanomolar
range concentraions of ouabaine. Remarkably, these occur as slow waves rather
than contin-uously. In the study cited, a secondary effect on a certain
transcription factor was noted as well. While the physiolog-ical role of
endogenous ouabain remains unsettled, the oc-currence of both temporal and
spatial waves and spikes of calcium signals is increasingly recognized, and
adds yet an-other layer of complexity to its role in cellular signalling.
Related Topics