Chapter: Biochemistry: Nucleic Acid Biotechnology
Cloning
Cloning
DNA
molecules containing covalently linked segments derived from two or more DNA
sources are called recombinant DNA.
(Another name for recombinant DNA is chimeric
DNA, named after the chimera, a monster in Greek mythology that had the
head of a lion, the body of a goat, and the tail of a serpent.) The production
of recombinant DNA was made possible by the isolation of restriction
endonucleases.
Using “Sticky Ends” to Construct Recombinant DNA
If
samples of DNA from two different sources are digested with the same
restriction enzyme and then mixed, in some cases the sticky ends that anneal to
Unfortunately,
when two different kinds of DNA are combined using restric-tion enzymes and DNA
ligase, relatively few product molecules are collected. Further experiments
with the DNA will require large amounts to work with, and this is made possible
by inserting the DNA into a viral or bacterial source. The virus is usually a
bacteriophage; the bacterial DNA typically is derived from a plasmid, a small circular DNA molecule
that is not part of the main circu-lar DNA chromosome of the bacterium. Using
DNA from a viral or bacterial source as one of the components of a recombinant
DNA enables scientists to take advantage of the rapid growth of viruses and
bacteria and to obtain greater amounts of the recombinant DNA. This process of
making identical copies of DNA is called cloning.
What is cloning?
The term
clone refers to a genetically
identical population, whether of organisms, cells, viruses, or DNA molecules.
Every member of the population is derived from a single cell, virus, or DNA
molecule. It is particularly easy to see how individual bacteriophages and
bacterial cells can produce large numbers of progeny. Bacteria grow rapidly,
and large populations can be obtained relatively easily under laboratory
conditions. Viruses also grow easily. We shall examine each of these examples
in turn.
A virus
can be considered a genome with a protein coat, usually consisting of many
copies of one kind of protein or, at most, a small number of different kinds of
proteins. The viral genome can be double-stranded or single-stranded DNA or
RNA. For purposes of this discussion, we confine our attention to DNA viruses
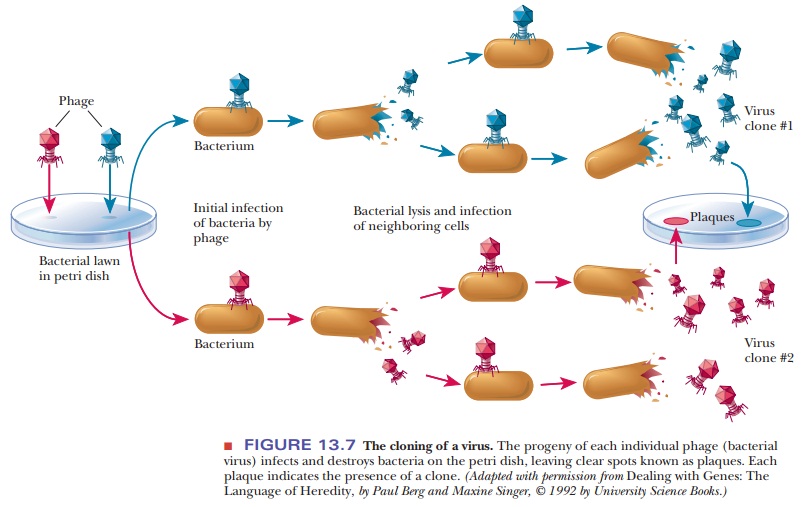
with double-stranded DNA. In the cloning of bacteriophages, a “lawn” of
bacteria covering a petri dish is infected with the phage. Each individual
virus infects a bacterial cell and reproduces, as do its progeny when they
infect and destroy other bacterial cells. As the virus multiplies, a clear
spot, called a plaque, appears on the
petri dish, marking the area in which the bacterial cellshave been killed
(Figure 13.7). The plaque consists of the progeny viruses that are clones of
the original.
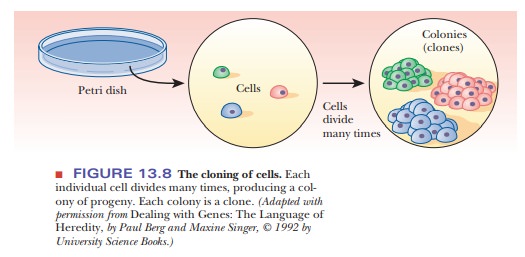
To clone
individual cells, whether from a bacterial or a eukaryotic source, a small
number of cells is spread thinly over a suitable growth medium in a dish.
Spreading the cells thinly ensures that each cell will multiply in isolation
from the others. Each colony of cells that appears on the dish will then be a
clone derived from a single cell (Figure 13.8). Because large quantities of
bacteria and bacte-riophages can be grown in short time intervals under
laboratory conditions, it is useful to introduce DNA from a larger,
slower-growing organism into bacteria or phages and to produce more of the
desired DNA by cloning. If, for example, we want to take a portion of human
DNA, which would be hard to acquire, and clone it in a virus, we can treat the
human DNA and the virus DNA with the same restriction endonuclease, mix the
two, and allow the sticky ends to anneal. If we then treat the mixture with DNA
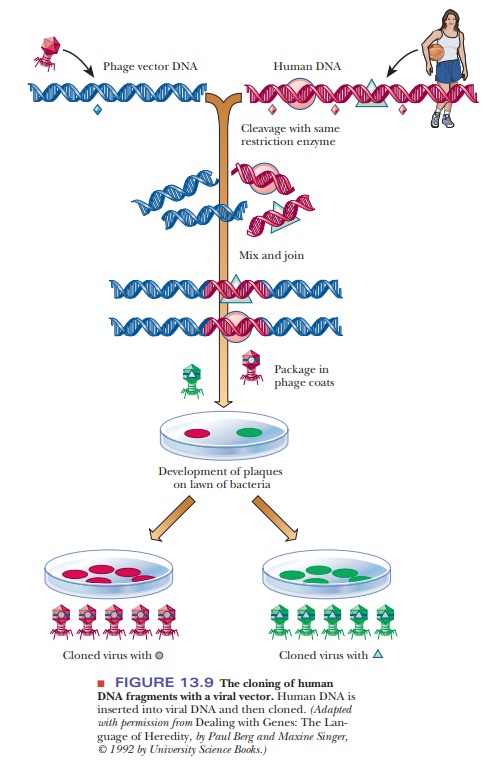
ligase, we have produced recombinant DNA. To clone it, we incorporate the
chimeric DNA into virus particles by adding viral coat protein and allowing the
virus to assemble itself. The virus particles are spread on a lawn of bacteria,
and the cloned segments in each plaque can then be identi-fied (Figure 13.9).
The bacteriophage is called a vector,
the carrier for the gene of interest that was cloned. The gene of interest is
called many things, such as the “foreign
DNA,” the “insert,” “geneX,” or
even “YFG,” for “your favorite gene.”
What are plasmids?
The other principal vector is a plasmid-bacterial DNA that is not part of the main circular DNA chromosome of the bacterium.
This DNA, which usually exists
as a closed circle, replicates independently of the main bacterial genome and
can be transferred from one strain of a bacterial species to another by cell-to-cell
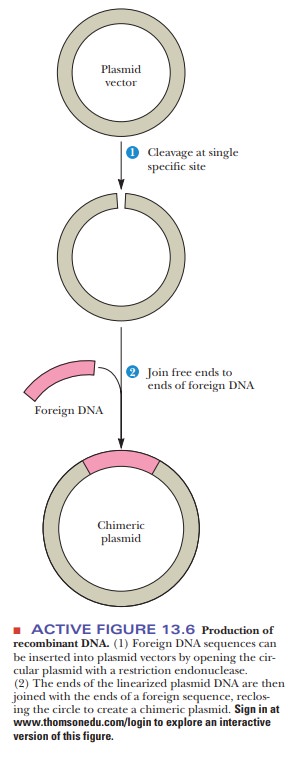
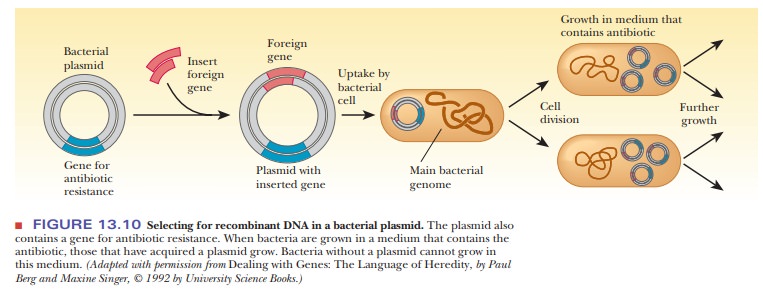
contact. The foreign gene can be inserted into the plasmid by the successive
actions of restriction endonucleases and DNA ligase, as was seen in Figure
13.6. When the plasmid is taken up by a bacterium, the DNA insert goes along
for the ride (Figure 13.10). The bacteria that contain the DNA insert can then
be grown in fermentation tanks under conditions that allow them to divide
rapidly, amplifying the inserted gene many thousandfold.
While
the theory of cloning DNA into a plasmid is straightforward, there are several
considerations for a successful experiment. When bacteria take up a plasmid, we
say they have been transformed.
Transformation is the process whereby new DNA is incorporated into a host.
Bacteria are encouraged to take up foreign DNA by a couple of methods. One is
to heat-shock the bacteria at 42°C, followed by placing them on ice. Another is
to place them in an electric field, a technique called electroporation.
How are we to know which of the bacteria have taken up the plasmid? Because bacteria divide quickly, we would not want all the bacteria to grow-rather, only the ones that have the plasmid. This process is called selection. Each plasmid chosen for cloning must have some type of selectable marker that lets us know that the growing bacterial colonies contain the plasmid. These markers are usually genes that confer resistance to antibiotics. After transformation, the bacteria are plated on a medium containing the antibiotic to which the plas-mid carries resistance.
In that way, only the bacteria that took up the plasmids will grow
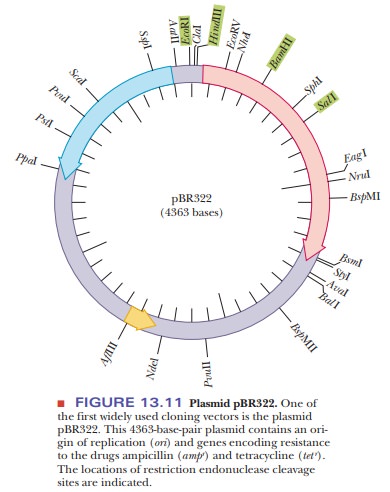
(Figure 13.10). One of the first plasmids used for cloning is pBR322 (Figure
13.11). This simple plasmid was created from a naturally occurring one found in
E. coli. Like all plasmids, it has an
origin of replication, so it can rep-licate independently of the rest of the
genome. It has genes that confer resis-tance to two antibiotics, tetracycline
and ampicillin. The genes are indicated tet
rand ampr. The
pBR322 plasmid has several restriction enzyme sites. Thenumber and location of
restriction sites is very important to a cloning experi-ment. The foreign DNA
must be inserted at unique restriction sites so that the use of restriction
enzymes opens up the plasmid at only one point. Also, if the restriction site
chosen is inside one of the selection markers, the resistance to the antibiotic
is lost upon insertion of the foreign DNA. This was, in fact, the original way
selection was done with plasmids. Foreign DNA was inserted using restriction
sites in the tetr gene. Selection was achieved
by noting the loss of the ability of bacteria to grow on a medium containing
tetracycline.
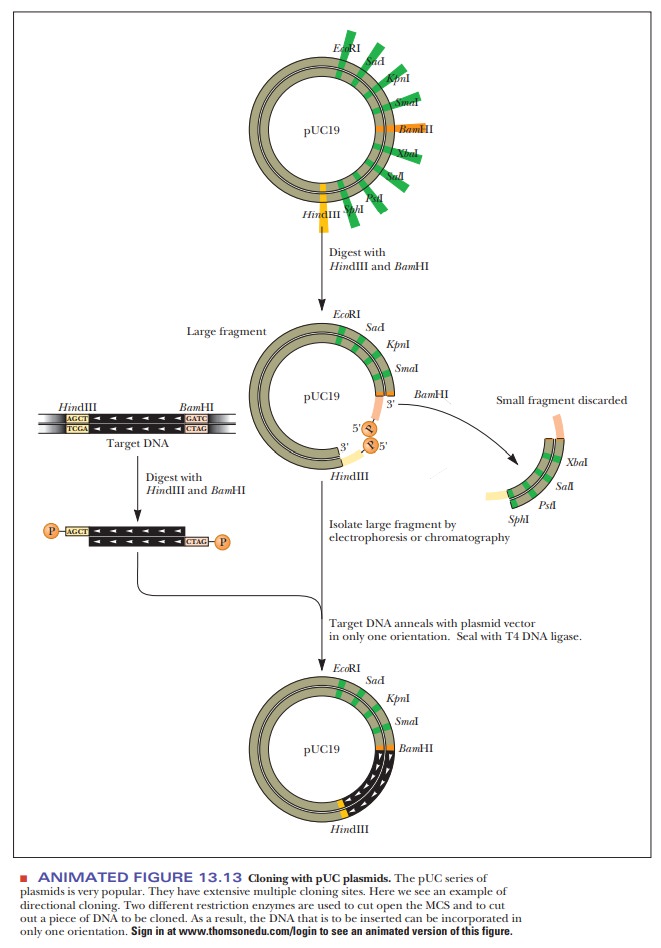
One of the early stumbling blocks to cloning was finding the right plasmid, one that had restriction sites that matched those enzymes needed to cut out the foreign DNA. As the technology to design plasmids improved, they were created with regions that had many different restriction sites in a small space. This region was called a multiple cloning site (MCS) or a polylinker (Figure 13.12). A popular cloning vector series is based on the pUC plasmids (Figure 13.13). The acronym pUC stands for universal cloning plasmid. Each of these cloning vectors has an extensive MCS, which helps solve another problem with cloning-the directionality of the inserted DNA.

Depending on what is to be done with the
cloned DNA, controlling its orientation in the vector may be important. If only
one restriction enzyme is used, such as BamH1,
then the foreign DNA can enter the plasmid in either of two directions.
However, if the foreign DNA is cut out of its source at one end with BamH1 and at the other end with HindIII, and if these same two
restriction enzymes are used to open up the plasmid, then the ends match in
only one direction (Figure 13.13).
The use of the pUC plasmids also aids in the selection procedure. The older plasmids that were based on pBR322 had the shortcoming that the foreign DNA was inserted into the tetracycline resistance gene. This meant that the only way to spot bacteria that had taken up a plasmid that had also taken up the insert was that the bacteria would not grow on a medium containing tet-racycline. This lack of growth by the successful clone made it challenging to go back and find the proper bacterial colonies. The pUC plasmids, however, have a characteristic that alleviates this procedure-they contain the lacZ gene, which is the basis for a selection technique called blue/white screening. The lacZ gene codes for theα-subunit of the enzymeβ-galactosidase, which is used to cleave disaccharides, such as lactose.
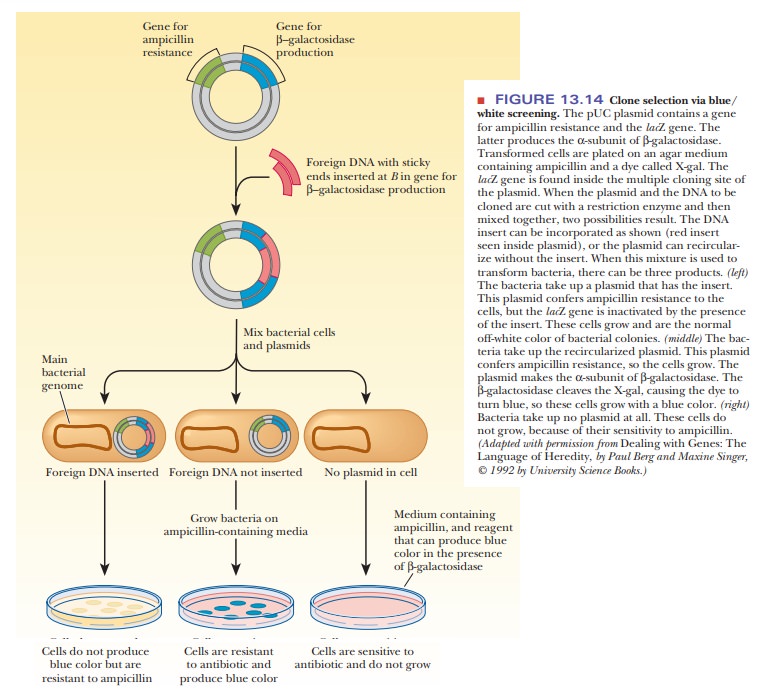
The MCS is located inside the lacZ gene, so that, when foreign DNA is
inserted, it inactivates the gene. Figure 13.14 shows how this characteristic
is useful. The pUC plasmid is cut open in its MCS by restriction enzymes, and
the foreign DNA (red) is cut out of its source with the same enzymes. These are
then combined and joined together with DNA ligase to yield two products in the
ligation reaction. The desired product is the plasmid that now contains the
foreign DNA. The other is a plasmid that has reclosed upon itself without the
inserted DNA. This type is much less common when two different restriction
enzymes are used to open the plasmid, but it still occurs infrequently. When
this mixture is used to trans-form the bacteria, there are three possible
products: (1) bacteria that took up the plasmid with the insert, (2) bacteria
that took up the plasmid without the insert, and (3) bacteria that took up no
plasmid at all. The mixture of bacteria from the transformation is plated on a
medium containing ampicillin and a dye, such as X-gal. β-Galactosidase hydrolyzes a bond in the X-gal
molecule, turning it blue. The bacteria to be transformed are mutants that make
a defec-tive version of the β-galactosidase that lacks the α-subunit. If the bacteria
take up no plasmid at all, they lack the ampicillin-resistance gene and do not
grow. If the bacteria take up a plasmid lacking the insert, they have a
functional lacZ gene and produce the α-subunit of β-galactosidase. These
colonies produce active β-galactosidase, which cleaves the dye, X-gal.
These colonies grow with a blue color. If the bacteria take up a plasmid that
contains the insert, the lacZ gene is
inactivated. These colonies are the off-white color of normal bacterial
colonies on agar.
Summary
Cloning refers to creating a genetically identical population.
DNA can be combined by using restriction
enzymes that create sticky ends in the DNA. This recombinant DNA has a target
DNA sequence that the experimenter is interested in.
The target DNA sequence is carried in some type of vector, usually
a bac-terial plasmid or a virus.
The target DNA sequence is inserted into a host
organism, and the natu-ral doubling time of the organism is used to create many
copies of the target DNA sequence.
Organisms
that are carrying the target DNA are identified through a pro-cess called
selection. Selection often involves antibiotic resistance.
Related Topics