Chapter: Basic & Clinical Pharmacology : The Gonadal Hormones & Inhibitors
Androgen Suppression & Antiandrogens
ANDROGEN SUPPRESSION &
ANTIANDROGENS
ANDROGEN SUPPRESSION
The
treatment of advanced prostatic carcinoma often requires orchiectomy or large
doses of estrogens to reduce available endog-enous androgen. The psychological
effects of the former and gynecomastia produced by the latter make these
approaches unde-sirable. As noted, the GnRH analogs such as goser-elin,
nafarelin, buserelin, and leuprolide acetate produce effective gonadal
suppression when blood levels are continuous rather than pulsatile.
ANTIANDROGENS
The
potential usefulness of antiandrogens in the treatment of patients producing
excessive amounts of testosterone has led to the search for effective drugs
that can be used for this purpose. Several approaches to the problem,
especially inhibition of synthesis and receptor antagonism, have met with some
success.
Steroid Synthesis Inhibitors
Ketoconazole, used primarily in the treatment of fungal disease,is an
inhibitor of adrenal and gonadal steroid synthesis, as described. It does not
affect ovarian aromatase, but it reduces human placental aromatase activity. It
displaces estradiol and dihydrotestosterone from sex hormone-binding protein in
vitro and increases the estradiol:testosterone ratio in plasma in vivo by a
different mechanism. However, it does not appear to be clinically useful in
women with increased androgen levels because of the toxicity associated with
prolonged use of the 400–800 mg/d required. The drug has also been used
experimentally to treat prostatic carcinoma, but the results have not been
encouraging. Men treated with ketoconazole often develop reversible
gyneco-mastia during therapy; this may be due to the demonstrated increase in
the estradiol:testosterone ratio.
Conversion of Steroid Precursors to Androgens
Several compounds have been developed that inhibit the 17-hydroxylation of progesterone or pregnenolone, thereby pre-venting the action of the side chain-splitting enzyme and the further transformation of these steroid precursors to active androgens. A few of these compounds have been tested clinically but have been too toxic for prolonged use. As noted, abiraterone, a newer 17-hydroxylase inhibitor, may prove to be clinically successful.
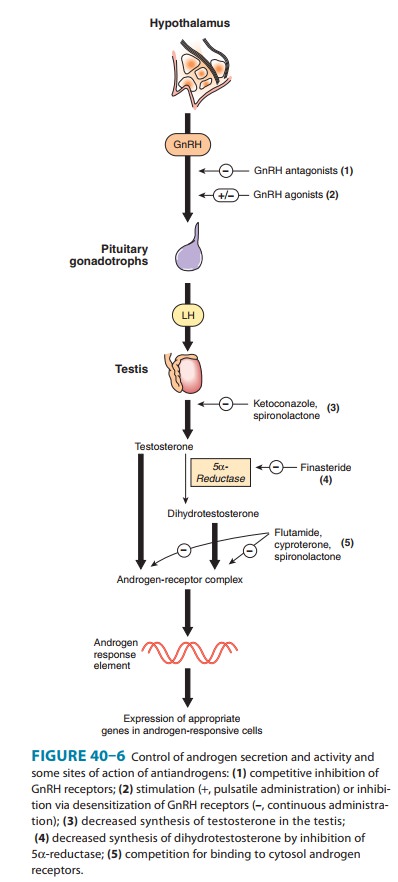
Since
dihydrotestosterone—not testosterone—appears to be the essential androgen in
the prostate, androgen effects in this and similar
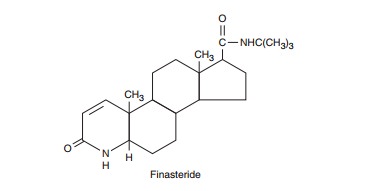
dihydrotestosterone-dependent tissues can be reduced by an inhibitor of 5α-reductase (Figure
40–6). Finasteride, a steroid-like
inhibitor of this enzyme, is orally active and causes a reduction in
dihydrotestosterone levels that begins within 8 hours after administration and
lasts for about 24 hours. The half-life is about 8 hours (longer in elderly
individuals). Forty to 50 percent of the dose is metabolized; more than half is
excreted in the feces. Finasteride has been reported to be moderately effective
in reduc-ing prostate size in men with benign prostatic hyperplasia and is
approved for this use in the USA. The dosage is 5 mg/d. Dutasteride is a similar orally active steroid derivative with a
slowonset of action and a much longer half-life than finasteride. The dosage is
0.5 mg daily. These drugs are not approved for use in women or children,
although finasteride has been used success-fully in the treatment of hirsutism
in women and early male pat-tern baldness in men (1 mg/d).
Receptor Inhibitors
Cyproterone and cyproterone acetate are
effective antiandrogensthat inhibit the action of androgens at the target
organ. The ace-tate form has a marked progestational effect that suppresses the
feedback enhancement of LH and FSH, leading to a more effec-tive antiandrogen
effect. These compounds have been used in women to treat hirsutism and in men
to decrease excessive sexual drive and are being studied in other conditions in
which the reduction of androgenic effects would be useful. Cyproterone acetate
in a dosage of 2 mg/d administered concurrently with an estrogen is used in the
treatment of hirsutism in women, doubling as a contraceptive pill; it has
orphan drug status in the USA.
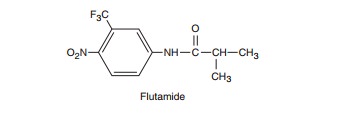
Flutamide, a
substituted anilide, is a potent antiandrogen thathas been used in the
treatment of prostatic carcinoma. Although not a steroid, it behaves like a
competitive antagonist at the androgen receptor. It is rapidly metabolized in
humans. It frequently causes mild gynecomastia (probably by increasing
testicular estrogen production) and occasionally causes mild reversible hepatic
toxicity. Administration of this compound causes some improvement in most
patients with prostatic carcinoma who have not had prior endocrine therapy.
Preliminary studies indicate that flutamide is also useful in the management of
excess androgen effect in women.
Bicalutamide and nilutamide are
potent orally active antian-drogens that can be administered as a single daily
dose and are used in patients with metastatic carcinoma of the prostate.
Studies in patients with carcinoma of the prostate indicate that these agents
are well tolerated. Bicalutamide is recommended for use in combination with a
GnRH analog (to reduce tumor flare) and may have fewer gastrointestinal side
effects than flutamide. A dos-age of 150–200 mg/d (when used alone) is required
to reduce prostate-specific antigen levels to those achieved by castration,
but, in combination with a GnRH analog, 50 mg/d may be ade-quate. Nilutamide is
approved for use following surgical castration in a dosage of 300 mg/d for 30
days followed by 150 mg/d.
Spironolactone, a
competitive inhibitor of aldosterone, also competes with dihydrotestosterone
for the andro-gen receptors in target tissues. It also reduces 17α-hydroxylase
activity, lowering plasma levels of testosterone and androstene-dione. It is
used in dosages of 50–200 mg/d in the treatment of hirsutism in women and
appears to be as effective as finasteride, flutamide, or cyproterone in this
condition.
Related Topics