Chapter: Biology of Disease: Disorders of the Immune System
The DiGeorge anomaly and the Wiskott Aldridge syndrome - Primary Immunodeficiency Disease
The DiGeorge anomaly and the Wiskott Aldridge syndrome
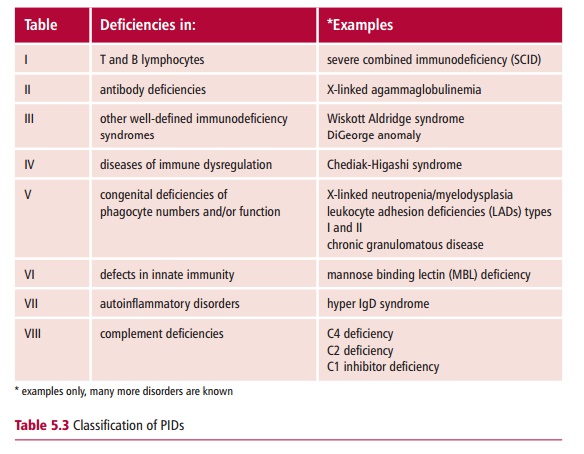
The third group of PIDs (Table 5.3) contains a number of well-defined immunodeficiency
syndromes, of which the DiGeorge anomaly and the Wiskott Aldridge syndrome are
well-known examples.
The DiGeorge anomaly (DGA) is a developmental
disorder involving organs that develop from the third and fourth pharyngeal
pouches of the embryo. It is associated with a deletion or partial monosomy of
chromosome 22 that results in a range of defects. Several different patterns of
inheritancehave been reported, including autosomal dominant and autosomal recessive.
Its incidence has been estimated to be between one in 20 000 to 66 000,
depending on the country.
The DGA is characterized by facial abnormalities,
hypoparathyroidism and hypocalcemia with symptoms of convulsions and tetany,
congenital heart disease that may be so severe as to be life threatening, and a
small under-developed or sometimes absent thymus that results in a profound
immuno-deficiency. Patients suffer severe and recurring viral and fungal
infections.
Indeed, the immunological defects are the second
commonest cause, after heart conditions, of death in DGA patients. The number
of circulating T lymphocytes is severely reduced leading to defects in
cell-mediated immunity. T cell proliferative responses to mitogens vary in DGA
patients, such that they can be classified either as partial or complete. In
the former, proliferation is reduced but in the latter it is completely absent.
The absence of helper T lymphocytes reduces antibody production, so that
antibacterial immunity may also be compromised, even though the number of
circulating B lymphocytes is normal.
A diagnosis of DGA is based on the cardiac
malformations, hypopara-thyroidism resulting in hypocalcemia and a small or
absent thymus. T lymphocytes in the circulation are reduced and the proliferative
response to mitogens is impaired. Fluorescence in situ hybridization (FISH) has been used to detect deletions in
chromosome 22 in the majority of patients (Margin
Note 5.1). Other syndromes,
without any apparent genetic link, but which have known environmental causes,
bear some resemblance to DGA. One example is fetal alcohol syndrome, which
results from prolonged exposure to alcohol during fetal development. Children
with fetal alcohol syndrome also show the characteristic facial features associated
with DGA.

Attempts have been made to treat the immunological
deficit in DGA with thymus transplants, although results have been variable.
The associated hypocalcemia is treated with calcium and vitamin D supplements,
while cardiac malformations must be rectified surgically. The prognosis for
patients with DGA is variable and depends mostly on the degree of
cardiovascular abnormality. For patients with severe cardiac problems it is
poor, with a mortality rate of over 80% at the age of six months.
The Wiskott Aldridge syndrome (WAS) arises from
mutation in the WAS gene, which was
identified on the short arm of the X chromosome in 1994. The gene codes for the
cytoskeletal protein sialophorin, found in lymphocytes and platelets, that is
involved in the assembly of actin filaments. The incidence of WAS is
approximately one per 250 000 male births.
The syndrome is characterized by decreased levels of
IgM but often with increased production of IgE and IgA. In the early stages, T
and B cell numbers in the blood are normal. Since IgM is the prevalent antibody
in immune responses to bacterial polysaccharides, there is an increased
incidence of infections with encapsulated bacteria. Sufferers may also develop
eczema. Blood platelets are small, short-lived and reduced in number, leading
to thrombocytopenia and increased bleeding times which may prove fatal . As WAS
progresses, there is a loss of both humoral and cell-mediated immunity and,
along with severe infections, there is also an increase in leukemia and lymphoid
tumors.
The treatment for WAS includes antibiotics for
infections and platelet transfusions to prevent bleeding. Immunoglobulin
replacement therapy may also be given to provide some protection against
infection. Bone marrow transplants have been successful in some cases.
Unfortunately the prognosis for WAS sufferers is poor, with death commonly
occurring before the age of four years usually from severe infection and
bleeding. Genetic counseling is recommended for women who have had a child with
WAS. Detection of the abnormal gene in cells obtained by chorionic villus
sampling or amniocentesis allows a prenatal diagnosis, with the possibility of
terminating the pregnancy if the fetus is found to be affected.
Related Topics