Chapter: Basic Concept of Biotechnology : Tools and Techniques in Biotechnology
Next Generation Sequencing (NGS) - Techniques of Biotechnology and Innovations
NEXT GENERATION SEQUENCING (NGS)
Nucleic acid sequencing helps in determining the order of nucleotides found in a DNA or RNA molecule. The use of nucleic acid sequencing has increased exponentially. The first main venture into DNA sequencing was the Human Genome Project, a $3 billion endeavour which took 13-year-long period to be completed in 2003. The Human Genome Project was performed with Sanger sequencing, first-generation sequencing. Sanger sequencing, the chain-termination method, was developed in 1975 by Edward Sanger and was used for nucleic acid sequencing for the successive two and a half decades (Sanger et al., 1977).
Demand for cheaper and faster sequencing methods has enhanced greatly since the completion of the first human genome sequence which lead to the development of second-generation sequencing methods, or next generation sequencing (NGS). NGS platforms execute enormous parallel sequencing and millions of fragments of DNA from a single sample are sequenced in unison. Massive parallel sequencing technology facilitates an entire genome tobe sequenced in less than one day. In the previous decade, numerous NGS platforms have been developed to provide low-cost and high-throughput sequencing. The Illumina MiSeq, The Life Technologies Ion Torrent Personal Genome Machine (PGM) and other NGS platforms has made sequencing available to more no. of labs. Resultantly, the volume of research and clinical diagnostics being performed with the help of nucleic acid sequencing is rapidly increasing.
Overview of the methodology
The Ion Torrent PGM and the Illumina MiSeq have a common base methodology that includes template preparation, sequencing and imaging, and data analysis. An overview of thesequencing methodologies is provided in Figure 12.
Template preparation
This step consists of construction of a library of DNA or complementary DNA and amplification of that library. Sequencing libraries are constructed by fragmenting the DNA or cDNA sample and ligating adapter sequences i.e. synthetic oligonucleotides of a known sequence onto the ends of DNA fragments. After construction, libraries are clonally amplified in preparation for the purpose of sequencing. The MiSeq utilizes bridge amplification for the formation of template clusters on a flow cell whereas the PGM utilizes emulsion PCR on the OneTouch system to increases single library fragments onto microbeads,
Sequencing and imaging
To acquire nucleic acid sequence from the amplified libraries, the MiSeq and the Ion Torrent PGM both rely on sequencing by the process of synthesis. The library fragments are used as template on which a new DNA fragment is synthesized. The sequencing comprises a cycle of washing and flooding of the fragments with the known nucleotides in a sequential order. As nucleotides integrate into theincreasing DNA strand, they are digitally recorded as sequence. The MiSeq and the PGM rely on a somewhat altered mechanism for detecting nucleotide sequence information. The MiSeq depends on the detection of fluorescence produced by the incorporation of fluorescently labelled nucleotides into the growing strand of DNA. By contrast, the PGM performs semiconductor sequencing that depends on the detection of pH changes caused by the release of a hydrogen ion upon the incorporation of a nucleotide into a growing strand of DNA.
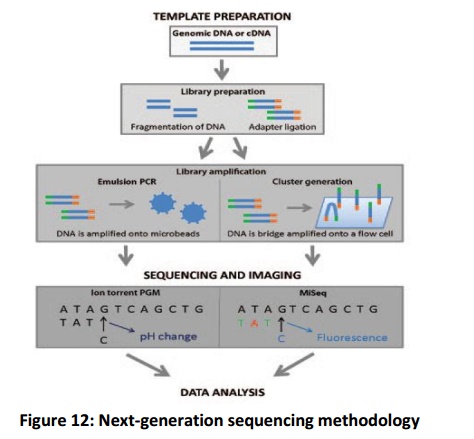
Data analysis
After completion of sequencing, raw sequence data undergo various analysis steps. A general data analysis for NGS data includes pre-processing of the data for removing adapter sequences and low-qualityreads, the mapping of the data to a reference genome. Analysis of the sequence includes a wide variety of assessments using bioinformatics, including detection of novel genes or regulatory elements, genetic variant calling for detection of SNPs or indels i.e. the insertion or deletion of bases and assessment of transcript expression levels. Analysis also includes the identification of somatic and germline mutation events that contributes to the correct diagnosis of a disease or genetic condition.
Applications
· Helps in comparative biology studies through whole genome sequencing of a wide variety of organisms.
· Sequencing of the human genome is being performed again to identify genes and regulatory elements implicated in pathological processes.
· Gene expression studies using RNA-Seq (NGS of RNA) have started replacing the use of microarray analysis which provides researchers the ability to analyse RNA expression in sequence form.
· In the fields of public health and epidemiology through the sequencing of bacterial and viral species to facilitate the identification of novel virulence factors.
NGS in practice
A) Whole-exome sequencing
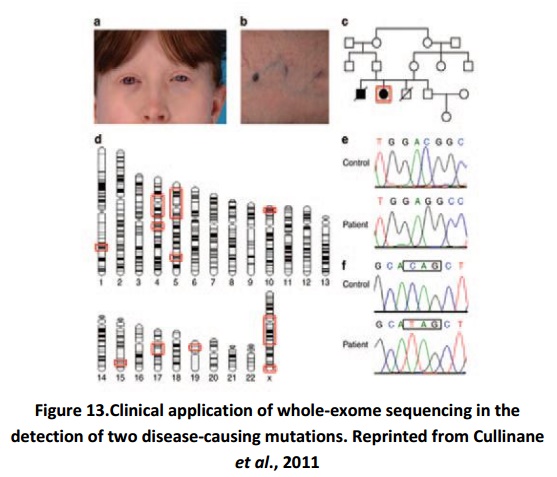
Exome sequencing is used extensively in the past several years for gene discovery research. Some gene discovery studies have resulted in the documentation of genes that are relevant to inherited skin diseases. It can also assist in the identification of disease-causing mutations where the particular genetic cause is not known.Figure 13establishes the direct effect that NGS in the correct diagnoses of a patient. It explains the practise of homozygosity mapping followed by whole-exome sequencing to recognize two disease- causing mutations in a patient with oculocutaneous albinism and congenital neutropenia. Figure 13a and 13b show the phenotypic traits common to oculocutaneous albinism type 4 and neutropenia detected in this patient. Figure 13c is a pedigree of the family of a patient, both the affected and unaffected individuals. The ideogram i.e. the graphic chromosome map in figure 13d stresses the regions of genetic homozygosity. These areas were acknowledged by single-nucleotide-polymorphism array analysis and were believed to be probable locations for the disease-causing mutation(s). Figures 13e and 13f exhibit chromatograms for the two disease-causing mutations recognised by whole-exome sequencing. Figure 13e portrays the mutation in SLC45A2, and Figure 12f shows the mutation in G6PC3. This case depicts the valuable role that NGS can play in the precise analysis of an individual patient who shows different symptoms with an unrevealed genetic cause.
B) Targeted sequencing
Targeted sequencing of specific genes or genomic regions is preferred when a suspected disease or condition has been identified. It is more affordable, reduces sequencing cost and time, and yields much higher coverage of genomic regions of interest. Sequencing panels are being developed for targeting hundreds of genomic regions known for hotspots for disease-causing mutations. Since, these sequencing panels target desired regions of the genome for sequencing; it eliminates the majority of the genome from analysis. The results of targeted sequencing of diseases help in decision making in many diseases therapeutically which includes different cancers for which the treatments should be cancer-type specific preferably.
Limitations
NGS is still too expensive for many lab conditions. Erroneous sequencing of homopolymer regions (spans of repeating nucleotides) on certain NGS platforms and short-sequencing read lengths (on average 200–500 nucleotides) can lead to sequence errors. Data analysis is sometimes time-consuming and requires knowledge of bioinformatics to harvest precise information from sequence data.
Related Topics