Chapter: Basic Concept of Biotechnology : Tools and Techniques in Biotechnology
In Situ Hybridization (ISH) - Techniques of Biotechnology and Innovations
IN SITU HYBRIDIZATION (ISH)
The localization of a fragment of specific nucleic acid in portion or section of tissue by hybridization with cDNA or RNA molecule as a probe is called as in situ hybridization (ISH). In general, the probes are labeled either with radioactive molecule or fluorescent molecules and their hybridization to DNA indicates the presence or absence of target DNA molecule in genome and their hybridization to RNA indicates the gene expression information.
Steps involved in ISH
1. Tissue preparation
2. Probe preparation
3. Hybridization and Visualization
1.Tissue preparation
Harvest the target tissue and rinse with 1x phosphate buffer saline (PBS). The dried tissues is immersed in freshly prepared 4% paraformaldehyde-0.1M sodium phosphate buffer pH7.4 and incubate at 4oC for 3hrs. The purpose of this step is not to completely fix the tissue but to harden and preserve the tissue. The complete fixation carried just before the hybridization which will fix all section equally. Later, tissues were immersed in 15% sucrose in 1x PBS (500mL sterile PBS, 75g "RNasefree" sucrose) for 3hrs to overnight at 4oC. Now, tissue is ready for sectioning and hence, tissues are embed in a matrix like parafilm in O.C.T blocks and used for sectioning. The tissues are sectioned on cryostat to 5-10 µm thickness, thaw-mounted onto coated slides and immediately flash frozen (-70oC).
2. Hybridization
The frozen tissues are mounted on slides and thawed for 5 min at 50oC this enhances the tissue retention on slide till the last step of the process. The dried slides are dipped in 4% paraformaldehyde (200mL 0.5M NaPO4, pH 7.4, 800mL DEPC H2O, Heat to 70°C with stirring on hot plate in fume hood Add 40g Paraformaldehyde) for 10 min at 4oC, this ensures the equally fixing of tissue on slide and it permanent retention of tissue on slides. The paraplast coated to tissue is removed by rinsing the tissue with xylene solution for 10 min. the slides are dipped in 100% ethanol for 15 mins or until strikes of xylene removed from the slide. These tissues are hydrated with series concentration of ethanol 95%, 85%, 70%, 50%, 30%, 15% and H2O. Repeat the repeat the step until strikes go away and tissue is hydrated. Incubate the slide in 0.2N HCL for 20 min at room temperature to denature the proteins and nicks the DNA. Later the slides are incubating in 2x SSC (1x SSC- 150 mM NaCl, 15 mM sodium acetate, pH7.0) 70°C, for 15 min and wash the slides with 1x PBS for 2 min at room temperature. Before hybridization, to increase the probe penetration, the tissue membrane softens by treating the tissue with 1.0 µg/mL of proteinase K for 10 min at 37oC. Wash the slides with sterile water for couple of times and rinse with 1x PBS. Further protease action inhibited by incubating the slides in 2mg/mL glycine in 1X PBS for 2 min at room temperature. Further, the slides are rinsed with 1x PBS for 2 times for 2 min. The dried slides are kept in hybridization box covered with 4x SSC buffer and 50% formamide saturated wick. Initially the pre-hybridization carried out where, 100 µl of hybridization buffer (forriboprobe hybridization-10 mM DTT, 0.3M NaCl, 20mM Tris pH 8.0, 5mM EDTA, 1x Denhardt, 10% Dextran sulphate, 50% formamide; for oligomer hybridization 10 mM DTT, 1x Denhardt, 5x SSC, 100 µg/mL of ssDNA, 100 µg/mL tRNA, 10% Dextran sulphate, 20% formamide) spotted on the tissue. The process goes for 3hrs at 42oC and later hybridization begins by adding the 20 µl of probe solution. The hybridization is carried out at 55oC for overnight incubation and next day, the slides are rinsed twice for 10 min with 2x SSC containing 10 mM β- mercaptoethanol and 1 mM EDTA at room temperature. Finally the tissue sections are dehydrated with gradient concentrated alcohol (15%, 30%, 50%, 70%, 85%, 95%, and 100%) containing 0.3M ammonium acetate and after bring it can be stored at room temperature. The hybridized probe can be visualized by epifluorescence microscopyfor fluorescent labelled probes and autoradiography used to observe the radioactive labelled probes.
3. Suppression subtractive hybridization (SSH)
SSH is a transcription profiling tool that aids in identifying the differentially expressed genes between two different (healthy and diseased) samples. The data explains type of genes that are differentially expressed in various biochemical pathways, regulatory pathways, signalling pathways, defense systems and adaptability. Apart from this, it co-relates the inter-relationship between identified genes, clustering of genes and understanding the cell behaviour at the molecular level. In SSH, mRNA population of one sample will be subtracted in mRNA population of other sample and differentially expressed mRNA copies are enriched. Usually the samples will be two counteracting samples. The sample in which genes are expected to express differentially is used as tester and its counteracting partner is driver. The SSH can be used in two different ways (i) forward (ii) reverse subtraction and both give different information such as upregulated and downregulated genes respectively. The technique explored in different biological system to identify andstudy gene expression pattern under different conditions (Diatchenko etal., 1996).
Expression of luxury genes depends upon conditions, confronting timings and type of host confronting. Hence, it is essential to consider these parameters to discover differentially expressed genes in tissue/cell. The SSH performed by preparing the cDNA from special condition (tester) and it will be subtracted in cDNA of its respectivecontrol (driver) condition (non-confronting sample). In the beginning step, the tester cDNA is divided into two groups (pool A and pool B) and each group ligated with one type of adaptor. In first round of hybridization, the driver cDNA allowed to hybridize with both the pool of cDNA separately. During the process the common genes between the tester and driver samples will get hybridize. In next round of hybridization, the product of first hybridization (pool A and pool B) mixed without any denaturation, allowing the hybridization to take place between unhybridized cDNA of pool A with unhybridized cDNA of pool B. During the process, the differentially expressed genes will get hybridized and these molecules carry two different adaptors at their ends. When this sample is subject to PCR with the adaptors specific primers than, the differentially expressedgenes will (because they carry two different adaptors at their ends) amplify and enriched with differentially expressed genes only. As the technique facilitating to discover the new genes and understanding their role in spatial condition, it is gaining more importance in research field. The SSH involves series of steps starting from, cDNA synthesis to amplification of differentially expressed gene and briefly described below (Fig. 4).
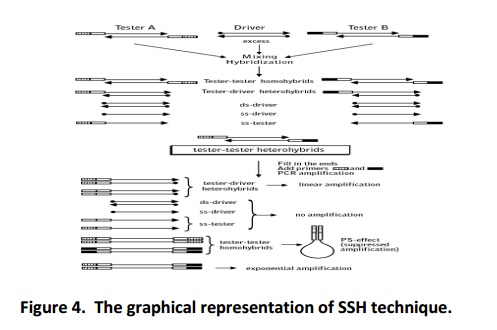
Methodology
Methodology mentioned here is based on the work of Diatchenko et al. (1996) and SSH kit protocol of TAKARA, USA. The 2 μgof poly A+ RNA is required concentration for good quality and quantity preparation of cDNA. The primer specific to poly A+ tail (10 μM ) is mixed and incubate for 2 min at 70oC which will allow the denature the soiled RNA molecule and annealing of primer to their location. Cool the tubes on ice for 2 min, the template hybridized to primer is ready for cDNA synthesis. Later, incubate the reaction with reverse transcriptase (100 units/μl, 1 μl/reaction), dNTP mix (10 mM each), 1X buffer and sterile water for 1.5 h at 42oC. Terminate the reaction by placing the tubes on ice and immediately proceeded for second strand cDNA synthesis. The RNA:DNA hybrid is denatured and remove the RNA molecule by RNase H enzyme (2 h incubation) and second strand is synthesised by adding the T4 DNA polymerase (6 U) per reaction. Incubate the tubes (in thermal cycler) at 16oC for 30 min, later the second strand synthesis was terminated by adding 4 µl of 20X EDTA/Glycogen mix and synthesised cDNA is precipitated and isolated by DNA purification method. The pellet is dissolve in sterile (43.5 µl) of water. The prepared double strand cDNA is digested with 1X of Rsa I enzyme as per the standard digestion procedure. It creates the blunt end double strands. Purify the digested short fragment by phenol: chloroform: iso amylalcohol method. The digested tester cDNA molecules is divide in to two group and each group attached with one type of adaptor with the help of T4 DNA ligase.
The adaptor ligated molecules are ready to hybridize with RsaI digested control sample cDNA molecules. Total two rounds of hybridization is carried out viz., (1) in first round of hybridization the adaptor ligated tester cDNA molecules of both the pools are mixed with digested control cDNA molecule individually in separate tubes. (2) The product of the first round hybridized pool B sample is mixed with is mixed product of the first round hybridized pool A with extra control driver cDNA molecules. The non-luxury genes will get hybridize in first round of hybridization and luxury genes with two different adaptors will hybridize in second round. During the first round of hybridization, the denaturation of sample is carried at 98oC for 1.5 min and whereas, in second round of hybridization the denaturation will not be followed. For first round of hybridization 8 h of incubation at 68oC and for second round of hybridization, the little longer time is required (10-12 h) at 68oC. These molecules further amplified with two rounds of PCR with primer specific to adaptor sequence. Hence, the cDNA molecule those carry both the types of adaptor will get amplify exponentially and the product will be enriched with only differentially expressed genes.
The 1 µl of each subtracted cDNA (needed to be diluted) of sample and their corresponding unsubtracted cDNA aliquot (this sample is collected before the subtraction) are subjected for the PCR with PCR primer 1. Initially, the tubes were incubated at 75oC for 5 min in a thermal cycler to extend the adaptors.In continuation of this incubation step, PCR program will commence (94oC for 20 sec, 94oC for 10 sec, 66oC for 30 sec and 72oC for 1.5 min. Dilute this product in 1:10 ratio and use 1 μl for second round of PCR. The nested primer used in second round of PCR it increase the specificity and only differentially expressed genes will exponentially amplify, the PCR program is 94oC for 10 sec, 68oC for 30 sec and 72oC for 1.5 min. The amplified product is stored at -20°C and used for sequencing.
Related Topics