Chapter: Basic Concept of Biotechnology : Tools and Techniques in Biotechnology
Isolation of Nucleic Acid (DNA) - Techniques of Biotechnology and Innovations
ISOLATION OF NUCLEIC ACID (DNA)
Fundamentally all living organisms are either unicellular or multicellular, which can grow, reproduce, process the information, respond to stimuli and process the several other chemical /biological/physical process. These all processes are governed by genetic information stored in the genome of an organism. Either prokaryotic of eukaryotic organism’s their genome is made of deoxyribonucleic acid (DNA). DNA is duplex of polynucleotide strands and it consisting of genes, promoter non-genic region and regulatory elements. To study this information and to characterize the genome of an organism, it is essential to isolate the genome (DNA) from their respective organism.
The total genetic materials vary from organism to organism and within the genera also. The DNA isolation methods more or less remain the same irrespective of size of the genome. However, the predominant factors like age of the sample and tissue type influence the isolation efficiency. The matured sample yield poor DNA quality than tender sample because of accumulation of phenolics like compounds.
The DNA isolation can be done in three phases.
1) Cell lysis,
3) DNA precipitation.
General protocol is mentioned in Fig. 1.
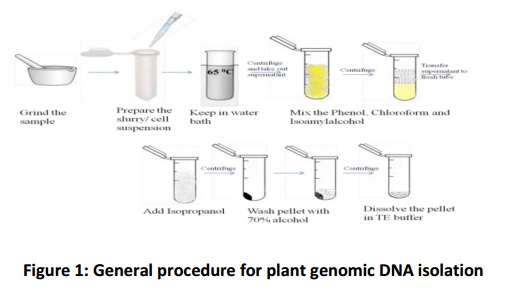
Phase 1) Breaking the cell wall and cell lysis
The purpose of this step is to break-down the cell wall of the cells. For good quality of DNA the sample should be young and fresh. In case of plants the 4- 6 tender leaves (0.5-1 gm) are plucked and plugged into ice, for bacterial culture the log phased grown (0.4-0.6 Optical density at 600 nm) and young mycelia from fungal strain are used for DNA isolation.
Phase 2) DNA extraction
This step is performed to extract the DNA from prepared slurry suspension in the previous step. Once the cell is crushed the cellular materials such as protein/ carbohydrate is released into extraction buffer. These compounds negatively affect further enzymatic reaction like restriction enzymes digestion and polymerase chain reaction (PCR). Hence, the removal of these compounds is essential and performed by adding the chloroform and isoamyl alcohol (24:1) or phenol, chloroform and isoamyl alcohol (25:24:1).
Phase 3) DNA precipitation
The isopropanol will cause the DNA strands to condense and become visible and it is denser than the solution that they are in. The reaction is allowed to happen in chilled condition and later precipitated by centrifuging it.
DNA isolation from plant leaves
1. The leaf samples are crushed in mortar and pestle by use of liquid nitrogen and β-mercaptoethanol is mixed with 500 µl of CTAB buffer to prepare the slurry.
2. The slurry collected in microcentrifuge tubes is incubated at 65oC for 30 min. During the process, the buffer components inactivate the DNase released from the cells.
3. The slurry is centrifuged at 10, 000 rpm for 10 min at 4oC to remove the cell debris and the aqueous layer containing clarified cell suspension is transferred in to a fresh tube.
4. The Chloroform and Isoamyl alcohol in 24:1 ratio is added to collect aqueous layer from previous step in an equal amount.
5. The tubes are centrifuged at 10,000 rpm for 10 in at 4oC and the aqueous layer is transferred to another tube without getting any contamination from middle and bottom layer.
6. An equal amount of pre chilled Isopropanol is mix with the collected aqueous layer of DNA solution from previous step.
7. It is mixed properly by inverting for couple of times and incubated for 30 min to overnight in deep freezer (-20oC).
8. Later, it is centrifuged at 10, 000 rpm for 10 min at 4oC and the collected pellet at the bottom of the tube is washed with 70% alcohol and it is spun for 2-5 min at 10, 000 rpm and the pellet is air dried.
9. The pellet is dissolved in 20-50 µl of TE buffer and stored in -20oC till future use.
DNA isolation from E. coli (DH5α) bacteria
1. In bacterial sample, the log phase cells (15 mL) are harvested and dissolved in 10mM Tris-Cl (3 mL) and 100 mM NaCl (3 mL).
2. The suspension is centrifuged at 10, 000 rpm for 10 min at 4oC and the pellet is re-dissolved in 2.5mL of TE buffer with 0.5 mL of
10mg/mL of lysozyme. The prepared cell suspensions are incubated at 37oC for 30 min by gently agitating occasionally.
3. 25 µl of RNase (10 mg/mL) is added and incubated at 37oC for 30 min.
4. 2.5 mL of SDS, which is prepared in 2 % TE buffer is added and incubated at 50oC for 45 min
5. Protein is removed by adding the 50 µl of Proteinase-K (20 mg/mL) and reaction is allowed to proceed at 55oC for 10 min.
6. The cleaved protein molecules are removed by mixing 6 mL of
Phenol with previously collected material, and then centrifuged at 10,000 rpm for 10 min at 4oC.
7. The aqueous phase is transferred to fresh tube and equal amount of phenol and chloroform in 1:1 ratio is added. The tube is centrifuged and the aqueous layer is collected. Chloroform and Isoamyl alcohol in ratio of 24:1 is added and the aqueous layer is collected from previous step.
8. The tubes are centrifuged at 10, 000 rpm for 10 min at 4oC.
9. 1/10th volume of 3 M Sodium Acetate is added to the aqueous layer collected from previous step.
10. After incubating in ice for 20 min equal volume of Isopropanol is added and incubated at room temperature for 5 min.
11. The pellet is centrifuged and washed in 100 µl of 70% Alcohol.
12. The pellet is dried in 25-30 µl in T10E1 and stored in -20oC for long term use.
DNA isolation from fungus (Trichoderma)
1. The fungal culture is incubated in potato dextrose broth (PDB) in 28oC for 3-5 days.
2. The 3 gm of fungal mat is harvested from edge of the growing fungal culture, homogenized in mortar, and pestle in 4 mL of 2% SDS for 5 min.
3. 6 mL of lysis buffer is added to the above supernatant.
4. To this mixture RNase (10 mg/mL) is added and incubated for 30 min in 37oC.
5. Equal amount of phenol, chloroform and isoamyl alcohol in
25:24:1 ratio is added. It is centrifuged at 10,000 rpm for 10 min at 4oC.
6. The supernatant is transferred to fresh tube and 1/10th volume of 3M sodium acetate and equal volume of isopropanol is added, it is mixed gently by inversion and incubated on ice for 30 min.
7. It is centrifuged at 10,000 rpm for 10 min at 4oC and the pellet is washed with 70% alcohol and the pellet is air dried.
8. The dried pellet is dissolved in 20-50 µl of TE buffer and stored in -20oC for future use.
DNA quality and quantity estimation
The purity and quantity of DNA is measured by spectro photometer or its purity is visualized on agarose gel. The DNA absorb the light wave length at 260 nm, the amount of absorbance is directly proportional to the amount of DNA present. The DNA sample is placed in quartz cuvette in order to read the OD. Based on absorbance coefficient the concentration of DNA is calculated. At A260 if the absorbance is 1 than concentration will be 50 µg/ mL and accordingly in other sample is calculated. For example, OD of DNA is 0.25 absorbance at A260 than concentration of DNA is 12.5 µg/ mL (0.25 × 50). At the mean time absorbance at 280 will give quantity of RNA in the sample. Hence, the A260/A280 gives the purity of DNA. If the ratio is 1.8 it indicate pure DNA, >1.8 shows presence of RNA and <1.8 shows protein contamination. It is visualized on horizontal agarose gel electrophoresis. In general, 1% of agarose is sufficient to visualize the genomic DNA. The gel is prepared by melting the 1 gm of agarose in 100 mL of TAE/TBE buffer. As the gel cools down the 3 µl of Ethidium Bromide (1 mg/mL) is dissolved and casted in the casting tray. The gel tray is immersed in buffer tank and ready for the DNA sample loading. The 2-3 µl of DNA ismixed with DNA gel loading dye (BPB/Orange dye) and load into wells formed in the gel tray. Electrophoresis of gel for 45min-60min and visualize under the UV light. The presence of intact high molecular weight band indicates the good quality of DNA and smeared band indicates degraded DNA. The good quality DNA further used for different purpose such as molecular mapping, isolation of gene and blotting.
Related Topics