Chapter: Pharmaceutical Biotechnology: Fundamentals and Applications : Formulation of Biotech Products, Including Biopharmaceutical Considerations
Microbiological Considerations
MICROBIOLOGICAL CONSIDERATIONS
Sterility
Most proteins are administered parenterally and have to be sterile. In general, proteins are sensitive to heat and other regularly used sterilization treatments; they cannot withstand autoclaving, gas sterilization, or sterilization by ionizing radiation. Consequently, sterilization of the end product is not possible. Therefore, protein pharmaceuticals have to be assembled under aseptic conditions, following the established and evolving rules in the pharmaceutical industry for aseptic manufacture. The reader is referred to standard textbooks for details (Halls, 1994; Groves, 1988; Klegerman and Groves, 1992).
Equipment and excipients are treated separately and autoclaved, or sterilized by dry heat (> 160deg. C), chemical treatment or gamma radiation to minimize the bioburden. Filtration techniques are used for removal of microbacterial contaminants. Prefilters remove the bulk of the bioburden and other particulate materials. The final “sterilizing” step before filling the vials is filtration through 0.2 or 0.22 mm membrane filters. Assembly of the product is done in class 100 (maximum 100 particles > 0.5 mm per cubic foot) rooms with laminar airflow that is filtered through high efficiency particulate air (HEPA) filters. Last but not least, the “human factor” is a major source of contamination. Well-trained operators wearing protective cloths (face masks, hats, gowns, gloves, or head-to-toe overall garments) should operate the facility. Regular exchange of filters, regular validation of HEPA equipment and thorough cleaning of the room plus equipment are critical factors for success.
Viral Decontamination
As recombinant DNA products are grown in microorganisms, these organisms should be tested for viral contaminants and appropriate measures should be taken if viral contamination occurs. In the rest of the manufacturing process, no (unwanted) viral material should be introduced. Excipients with a certain risk factor such as blood-derived human serum albumin should be carefully tested before use and their presence in the formulation process should be minimized.
Pyrogen Removal
Pyrogens are compounds that induce fever. Exogenous pyrogens (pyrogens introduced into the body, not generated by the body itself) can be derived from bacterial, viral or fungal sources. Bacterial pyrogens are mainly endotoxins shed from gramnegative bacteria. They are lipopolysaccharides. A general structure is shown in Figure 1. The basic, conserved structure in the full array of thousands of different endotoxins is the lipid A-moiety. Another general property shared by endotoxins is their high, negative electrical charge. Their tendency to aggregate and to form large units with MW of over 106 in water and their tendency to adsorb to surfaces indicate that these compounds are amphipathic in nature. They are stable under standard autoclaving conditions, but break down when heated in the dry state. For this reason equipment and container are treated at temperatures above 160deg. C for prolonged periods (e.g., 30 minutes dry heat at 250dg.C Pyrogen removal of recombinant products derived from bacterial sources should be an integral part of the preparation process. Ion exchange chromatographic procedures (utilizing its negative charge) can effectively reduce endotoxin levels in solution.
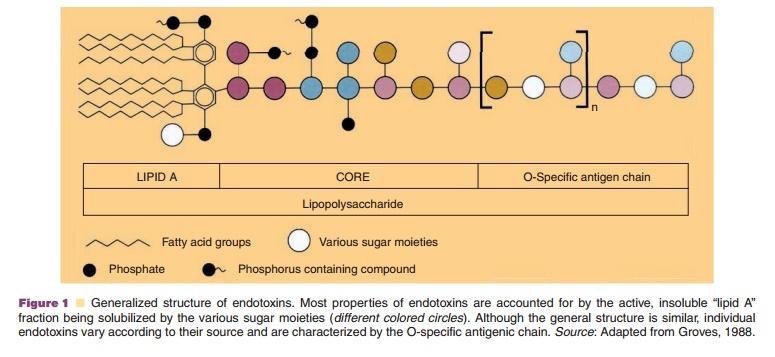
Excipients used in the protein formulation should be essentially endotoxin free. For solutions “Water for Injection” (compendial standards) is (freshly) distilled or produced by reverse osmosis.
The aggregated endotoxins cannot pass through the reverse osmosis membrane. Removal of endotoxins immediately before filling the final container can be accomplished by using activated charcoal or other materials with large surfaces offering hydrophobic interactions. Endotoxins can also be inactivated on utensil surfaces by oxidation (e.g., peroxide) or dry heating (e.g., 30 minutes dry heat at 250 C).
Related Topics