Chapter: Pharmaceutical Biotechnology: Fundamentals and Applications : Formulation of Biotech Products, Including Biopharmaceutical Considerations
Excipients Used in Parenteral Formulations of biotech Products
EXCIPIENTS USED IN PARENTERAL FOR MULATIONS OF BIOTECH PRODUCTS
In a protein formulation one finds, apart from the active substance, a number of excipients selected to serve different purposes. This process of formulation design should be carried out with great care to ensure therapeutically effective and safe products. The nature of the protein (e.g., lability) and its therapeutic use (e. g., multiple injection systems) can make these formulations quite complex in terms of excipient profile and technology (freeze-drying, aseptic pre-paration). Table 1 lists components that can be found in the presently marketed formulations. In the following sections this list is discussed in more detail.

Solubility Enhancers
Proteins, in particular those that are non-glycosylated, may have a tendency to aggregate and precipitate. Approaches that can be used to enhance solubility include selection of the proper pH and ionic strength conditions. Addition of amino acids such as lysine or arginine (used to solubilize tissue plasminogen activator, t-PA), or surfactants such as sodium dodecylsulfate to solubilize non-glucosylated IL-2 can also help to increase the solubility. Themechanism of action of these solubility enhancers depends on the type of enhancer and the protein involved and is not always fully understood.
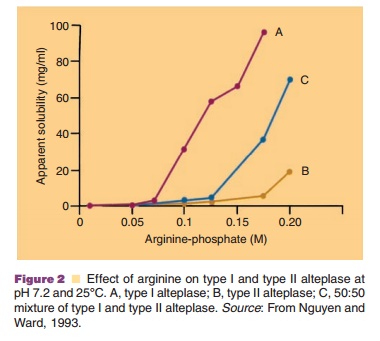
Figure 2 shows the effect of arginine concentra-tion on the solubility of t-PA (alteplase) at pH 7.2 and 25 C. This figure clearly indicates the dramatic effect of this basic amino acid on the apparent solubility of t-PA.
In the above examples, aggregation is physical in nature, i.e. based on hydrophobic and/or electrostatic interactions between molecules. However, aggrega-tion based on the formation of covalent bridges between molecules through disulfide bonds, and ester or amide linkages has been described as well (see Table 4). In those cases, proper conditions should be found to avoid these chemical reactions.
Anti-Adsorption and Anti-Aggregation Agents
Anti-adsorption agents are added to reduce adsorp-tion of the active protein to interfaces. Some proteins tend to expose hydrophobic sites, normally present in the core of the native protein structure when an interface is present. These interfaces can be water/air, water/container wall or interfaces formed between the aqueous phase and utensils used to administer the drug (e.g., catheter, needle). These adsorbed, partially unfolded protein molecules form aggregates, leave the surface, return to the aqueous phase, form larger aggregates and precipitate. As an example, the proposed mechanism for aggregation of insulin in aqueous media through contact with a hydrophobic surface (or water–air interface) is presented in Figure 3 (Thurow and Geisen, 1984).
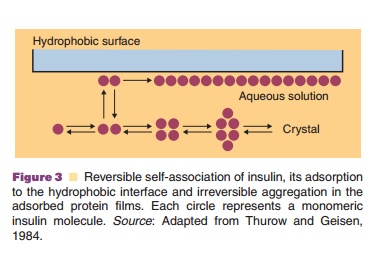
Native insulin in solution is in an equilibrium state between monomeric, dimeric tetrameric, and hexameric forms. The relative abundance of the different aggregation states depends on the pH, insulin concentration, ionic strength, and specific excipients (e.g., Zn2þ and phenol). It has been suggested that the dimeric form of insulin adsorbs to hydrophobic interfaces and subsequently forms larger aggregates at the interface. This explains why anti-adhesion agents can also act as anti-aggregation agents. Albumin has a strong tendency to adsorb to surfaces and is therefore added in relatively high concentrations (e.g., 1%) to protein formulations as an anti-adhesion agent. Albumin competes with the therapeutic protein for binding sites and supposedly prevents adhesion of the therapeutically active agent by a combination of its binding tendency and abundant presence.
Insulin is one of the many proteins that can form fibrillar precipitates (long rod-shaped structures with diameters in the 0.1 mm range). Low concentrations of phospholipids and surfactants have been shown to exert a fibrillation-inhibitory effect. The selection of the proper pH can also help to prevent this unwanted phenomenon (Brange and Langkjaer, 1993).
Apart from albumin, surfactants can also pre-vent adhesion to interfaces and precipitation. These molecules readily adsorb to hydrophobic interfaces with their own hydrophobic groups and render this interface hydrophilic by exposing their hydrophilic groups to the aqueous phase.
Buffer Components
Buffer selection is an important part of the formula-tion process, because of the pH dependence of protein solubility and physical and chemical stability. Buffer systems regularly encountered in biotech formula-tions are phosphate, citrate, and acetate. A good example of the importance of the isoelectric point (its negative logarithm is equal to pI) is the solubility profile of human growth hormone (hGH, pI around 5) as presented in Figure 4.
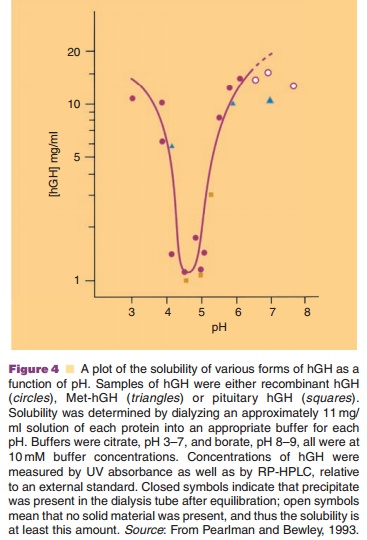
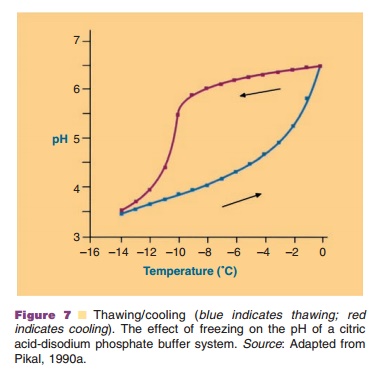
Even short, temporary pH changes can cause aggregation. These conditions can occur, for example during the freezing step in a freeze-drying process, when one of the buffer components is crystallizing and the other is not. In a phosphate buffer, Na2HPO4 crystallizes faster than NaH2PO4. This causes a pronounced drop in pH during the freezing step. Other buffer components do not crystallize, but form amorphous systems and then pH changes are minimized.
Preservatives and Antioxidants
Methionine, cysteine, tryptophan, tyrosine, and histi-dine are amino acids that are readily oxidized (Table 4). Proteins rich in these amino acids are liable to oxidative degradation. Replacement of oxygen by inert gases in the vials helps to reduce oxidative stress. Moreover, the addition of antiox-idants such as ascorbic acid or acetylcysteine can be considered. Interestingly, destabilizing effects on proteins have been described for antioxidants as well (Vemuri et al., 1993b). Ascorbic acid, for example, can act as an oxidant in the presence of a number of heavy metals.
Certain proteins are formulated in containers designed for multiple injection schemes. After admin-istering the first dose, contamination with microor-ganisms may occur and preservatives are needed to minimize growth. Usually, these preservatives are present in concentrations that are bacteriostatic rather than bactericidal in nature. Antimicrobial agents mentioned in the USP 29 are the mercury-containing phenylmercuric nitrate and thimerosal and p-hydro-xybenzoic acids, phenol, benzyl alcohol and chlor-obutanol (USP 29; Groves, 1988; Pearlman and Bewley, 1993). The use of mercury containing pre-servatives is presently under discussion (FDA, 2006).
Osmotic Agents
For proteins the regular rules apply for adjusting the tonicity of parenteral products. Saline and mono- or disaccharide solutions are commonly used. These excipients may not be inert; they may influence protein structural stability. For example, sugars and polyhydric alcohols can stabilize the protein structure through the principle of “preferential exclusion” (Arakawa et al., 1991). These additives (water struc-ture promoters) enhance the interaction of the solvent with the protein and are themselves excluded from the protein surface layer; the protein is preferentially hydrated. This phenomenon can be monitored through an increased thermal stability of the protein. Unfortunately, a strong “preferential exclusion” effect enhances the tendency of proteins to self-associate.
Shelf Life of Protein-Based Pharmaceuticals
Proteins can be stored (i) as an aqueous solution, (ii) in freeze-dried form, (iii) in dried form in a compacted state (tablet). Some mechanisms behind chemical and physical degradation processes have been briefly discussed.
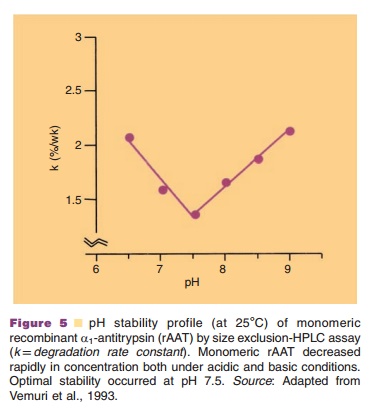
Stability of protein solutions strongly depends on factors such as pH, ionic strength, temperature, and the presence of stabilizers. For example, Figure 5 shows the pH dependence of a1-antitrypsin and clearly demonstrates the critical importance of pH for the shelf life of proteins.
Freeze Drying of Proteins
Proteins in solution often do not meet the preferred stability requirements for industrially produced phar-maceutical products (> 2 years), even when kept permanently under refrigerator conditions (cold chain). The abundant presence of water promotes chemical and physical degradation processes.
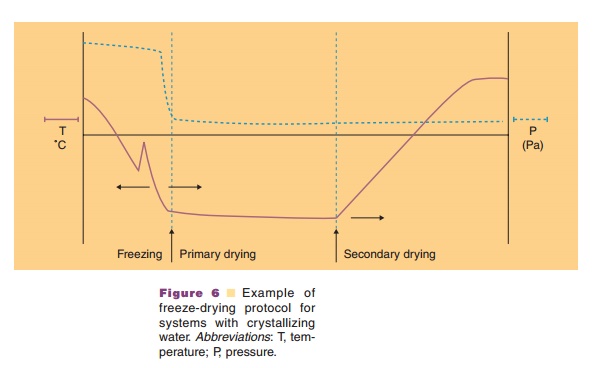
Freeze drying may provide the requested stability. During freeze-drying water is removed through sublimation and not by evaporation. Three stages can be discerned in the freeze-drying process: (i) a freezing step, (ii) the primary drying step, and (iii) the secondary drying step (Fig. 6). Table 2 explains what happens during these stages.
Freeze drying of a protein solution without the proper excipients causes, as a rule, irreversible damage to the protein. Table 3 lists excipients typically encountered in successfully freeze-dried protein products.


Freezing
In the freezing step (Fig. 6) the temperature of the aqueous system in the vials is lowered. Ice crystal formation does not start right at the thermodynamic or equilibrium freezing point, but supercooling occurs. That means that crystallization often only occurs when temperatures of –15 C or lower have been reached. During the crystallization step the temperature may temporarily rise in the vial, because of the generation of crystallization heat. During the cooling stage, concentration of the protein and excipients occurs because of the growing ice crystal mass at the expense of the aqueous water phase. This can cause precipitation of one or more of the excipients, which may consequently result in pH shifts (see above and Fig. 7) or ionic strength changes. It may also induce protein denaturation. Cooling of the vials is done through lowering the temperature of the shelf. Selecting the proper cooling scheme for the shelf, and consequently vial, is important as it dictates the degree of supercooling and ice crystal size. Small crystals are formed during fast cooling; large crystals form at lower cooling rates. Small ice crystals are required for porous solids and fast sublimation rates (Pikal, 1990a).
If the system does not (fully) crystallize but forms an amorphous mass upon cooling, the tem-perature in the “freezing stage” should drop below Tg, the glass transition temperature. In amorphoussystems the viscosity changes dramatically in the temperature range around the Tg: a “rubbery” state exists above and a glass state below the Tg.
At the start of the primary drying stage no “free and fluid” water should be present in the vials. Minus forty degrees Celsius is a typical freezing temperature before sublimation is initiated through pressure reduction.
Primary Drying
In the primary drying stage (Fig. 6) sublimation of the water mass in the vial is initiated by lowering the pressure. The water vapor is collected on a condenser, with a (substantially) lower temperature than the shelf with the vials. Sublimation costs energy (about 2500 kJ/gram ice). Temperature drops are avoided by
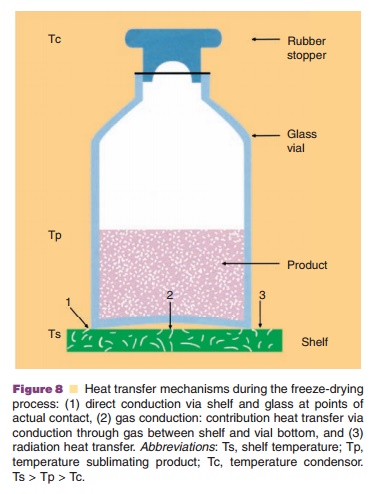
Heat is transferred to the vial through (i) direct shelf-vial contact (conductance), (ii) radiation, and (iii) gas conduction (Fig. 8). Gas conduction depends on the pressure: if one selects relatively high gas pressures, heat transport is promoted because of a high conductivity. But, it reduces mass transfer, because of a low driving force: the pressure between equilibrium vapor pressure at the interface between the frozen mass/dried cake and the chamber pressure (Pikal, 1990a).
During the primary drying stage one transfers heat from the shelf through the vial bottom and the frozen mass to the interface frozen mass/dry powder, to keep the sublimation process going. During this drying stage the vial content should never reach or exceed the eutectic temperature or glass transition temperature range. Typically a safety margin of 2 C to 5 C is used, otherwise the cake will collapse. Collapse causes a strong reduction in sublimation rate and poor cake formation. Heat transfer resistance decreases during the drying process as the transport distance is reduced by the retreating interface. With the mass transfer resistance (transport of water vapor), however, the opposite occurs. Mass transfer resistance increases during the drying process as the dry cake becomes thicker.
This situation makes it clear that parameters such as chamber pressure and shelf heating are not necessarily constant during the primary drying
process. They should be carefully chosen and adjusted as the drying process proceeds.
The eutectic temperature or glass transition temperature are parameters of great importance to develop a rationally designed freeze-drying protocol. Information about these parameters can be obtained by microscopic observation of the freeze-drying process, differential scanning calorimetry (DSC), or electrical resistance measurements.
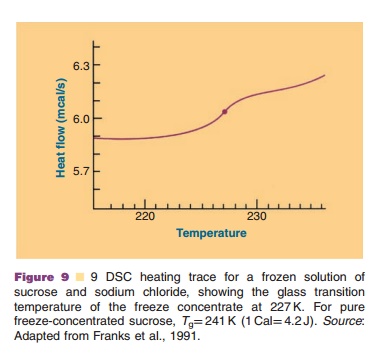
An example of a DSC scan providing informa-tion on the Tg is presented in Figure 9 (Franks et al., 1991). The Tg heavily depends on the composition of the system: excipients and water content. Lowering the water content of an amorphous system causes the Tg to shift to higher temperatures.
Secondary Drying
When all frozen or amorphous water that is non-protein and non-excipient bound is removed, the secondary drying step starts (Fig. 6). The end of the primary drying stage is reached when product temperature and shelf temperature become equal, or when the partial water pressure drops (Pikal, 1990a). As long as the “non-bound” water is being removed, the partial water pressure almost equals the total pressure. In the secondary drying stage the tempera-ture is slowly increased to remove “bound” water; the chamber pressure is still reduced. The temperature should stay all the time below the collapse/eutectic temperature, which continues to rise when residual water contents drop. Typically, the secondary drying step ends when the product has been kept at 20 C for some time. The residual water content is a critical, endpoint indicating parameter. Values as low as 1% residual water in the cake have been recommended.
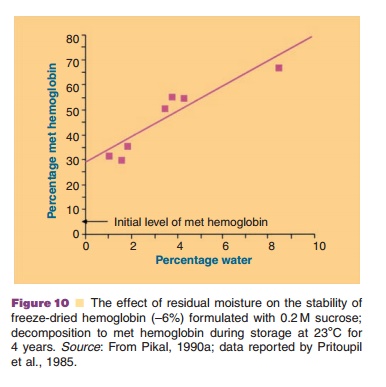
Figure 10 (Pristoupil, 1985; Pikal, 1990a) exemplifies the decreasing stability of freeze-dried hemoglobin with increasing residual water content.
When stored in the presence of reducing lyoprotectants such as glucose and lactose the Maillard reaction may occur: amino groups of the proteins react with the lyoprotectant in the dry state and the cake color turns yellow-brown. The use of non-reducing sugars such as sucrose or trehalose may avoid this problem.
Other Approaches to Stabilize Proteins
Compacted forms of proteins are being used for certain veterinary applications, such as sustained release formulations of growth hormones. The pellets should contain as few additives as possible. They can be applied subdermally or intramuscularly when the compact pellets are introduced by compressed air-powered rifles into the animals (Klegerman and Groves, 1992).
Related Topics