Chapter: Pharmaceutical Biotechnology: Fundamentals and Applications : Formulation of Biotech Products, Including Biopharmaceutical Considerations
Colloidal Particulate Carrier Systems for TargetedDelivery of Proteins: Nanotechnology at Work
Colloidal Particulate Carrier Systems for Targeted Delivery
of Proteins: Nanotechnology at Work
A wide range of carrier systems in the colloidal size range (diameters
up to a few micrometers) has been proposed for protein targeting. Examples are:
lipo-somes, biodegradable polycyanoacrylate nanoparti-cles, albumin
microspheres, polylactic acid microspheres, and low density lipoproteins (LDL).
Upon entering the bloodstream after IV injection, it is difficult for many of
these particulate systems to pass through epithelial and endothelial membranes
in healthy tissue, as the size cut-off for permeation through these
multilayered barriers is around 20 nm (excluding the liver, see above and Fig.
26). Parameters that control the fate of particulate carriers in vivo are
listed in Table 15.

As a rule, cells of the mononuclear phagocyte system (MPS), such as macrophages, recognize stable, colloidal particulate systems (< 5 mm) as “foreign body like structures” and phagocytose them. Thus, the liver and spleen, organs rich in blood circulation exposed macrophages, take up the majority of these particu-lates (Tomlinson, 1987; Crommelin and Storm, 1990). Larger (> 5 mm) intravenously injected particles tend to form emboli in lung capillaries on their first encounter with this organ.
Liposomes have gained considerable attention among the colloidal
particulate systems proposed for site-specific delivery of (or by) proteins.
Liposomes are vesicular structures based on (phospho)lipid bilayers surrounding
an aqueous core. The main component of the bilayer usually is
phosphatidylcho-line (Fig. 30).
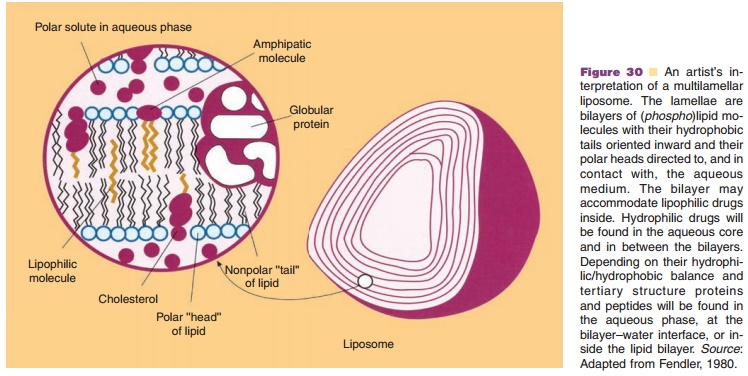
By selecting their bilayer constituents and one of the many preparation
procedures described, lipo-somes can be made varying in size between 30 nm
(e.g., by extrusion or ultrasonication) and 10 mm, and
charge (by incorporation negatively or positively charged lipid molecules), and
bilayer rigidity (by selecting special phospholipids or adding lipids such as
cholesterol). Liposomes can carry their payload (proteins) either in the lipid
core of the bilayer through partitioning, attached to the bilayer, or
physically entrapped in the aqueous phase. To make liposomes target site
specific, except for passive targeting to liver (Kupffer cells) and spleen
macro-phages, homing devices are covalently coupled to the outside bilayer
leaflet (Toonen and Crommelin, 1983). In Table 16 three relative advantages of
liposomes over other particulate systems are given.

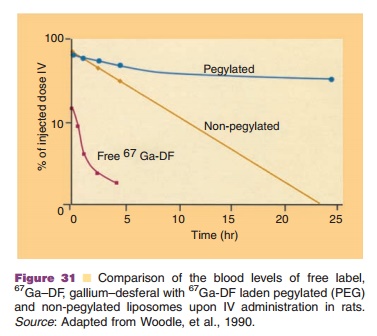
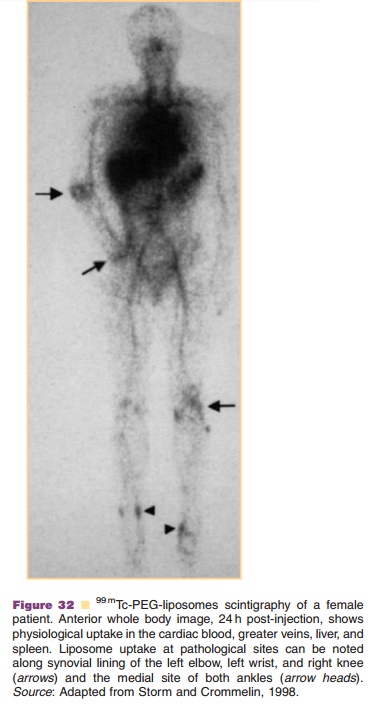
After injection “standard” liposomes stay in the blood circulation only for a short time. They are taken up by macrophages in liver and spleen, or they degrade by exchange of bilayer constituents with blood constituents. Liposome residence time in the blood circulation can be extended to many hours and even days, if PEG chains are grafted on the surface and stable bilayer structures are used (Fig. 31, see also Fig. 17). These long circulating liposomes apparently are able to escape macrophage uptake for prolonged periods of time and are sequestered in other organs than liver and spleen alone, e.g. tumors and inflamed tissues. In Figure 32 an example is shown of the use of mTc-labelled liposomes in the detection of inflam-mation sites in a patient.
The accumulation of protein-laden liposomes in macrophages (passive targeting) offers interesting therapeutic opportunities. Liposome encapsulated lymphokines and “microbial” products, such as interferon-a or muramyl tripeptide phosphatidyletha-nolamine (MTP-PE), respectively, can activate macrophages and enable them to kill micrometas-tases, or help to stimulate immune reactions. Moreover, reaching macrophages may help us to more effectively fight macrophage located microbial, viral or bacterial diseases than with our present approaches (Emmen and Storm, 1987; Crommelin and Schreier, 1994).
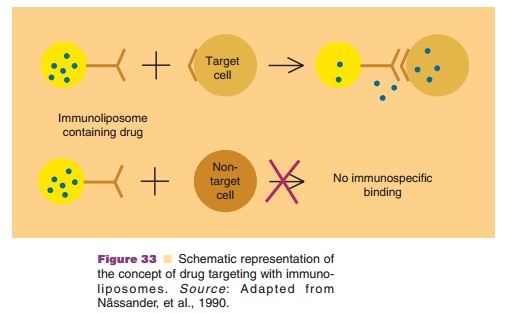
Several attempts have been made to sequester immunoliposomes (i.e.,
antibody (fragment)-liposome combinations) at predetermined sites in the body.
Here the aim is active targeting to the desired target site instead of passive
targeting to macrophages. The concept is schematically presented in Figure 33.
When designing immunoliposomes, antibodies or antibody-fragments are
covalently bound to the surface of liposomes through lipid anchor molecules
(Toonen and Crommelin, 1983). Non-PEGylated im-munoliposomes have poor access
to target sites outside the blood circulation after IV injection. The reason is
the high resistance against liposome pene-tration through the endothelial
lining at target sites and their relatively short circulation time (Fig. 26).
Therefore, target sites should be sought in the blood circulation (red blood
cells, thrombi, lymphocytes, or endothelial cells exposing under stress certain
adhe-sion molecules, e.g. ICAM-1, intercellular cell adhe-sion molecule)
(Vingerhoeds et al., 1994; Crommelin et al., 1995).
Other interesting target sites are those located in cavities, where one
can locally administer the drug-carrier combination. The bladder and the
peritoneal cavity are such cavities. These cavities can be the sites where the
diseased tissue is concentrated. For instance, with ovarian carcinomas the
tumors are confined to the peritoneal cavity for most of their lifetime. After IP
injection of immunoliposomes directed against human ovarian carcinomas in
athy-mic, nude mice, a specific interaction between immunoliposomes and the
human ovarian carcinoma was observed (Na¨ssander and colleagues) (Fig. 34).
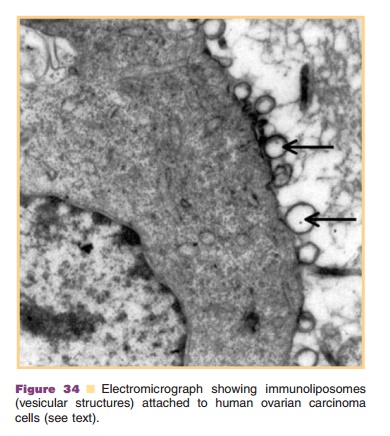
A new generation of liposomes is under devel-opment that combines PEG
coating (long circulation characteristic) and antibody coating (targeting).
Attaching an immunoliposome to target cells usually does not induce a
therapeutic effect per se. After establishment of an immunoliposome–cell
inter-action the protein drug has to exert its action on the cell. To do that,
the protein has to be released in its active form. There are several pathways
proposed to reach this goal (Fig. 35) (Peeters et al., 1987).
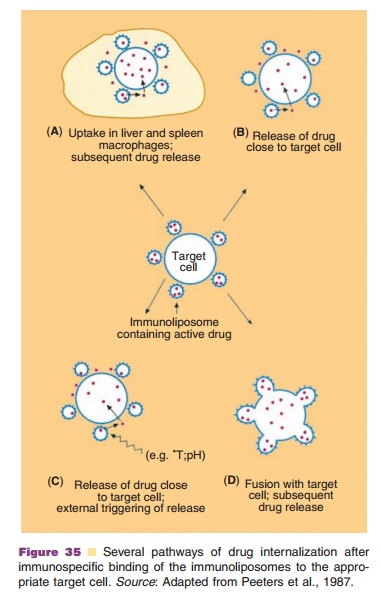
When the immunoliposome–cell complex en-counters a macrophage, the cells plus adhering liposome are probably phagocytosed and enter themacrophage (option A). Subsequently, the liposome-associated protein drug can be released. As this will most likely happen in the “hostile” lysosomal envir-onment, little intact protein will become available. In the situation depicted in Figure 35, option B, the drug is released from the adhering immunoliposomes in the close proximity of the target cell. In principle, release rate control is achieved by selecting the proper liposomal bilayers with delayed or sustained drug release characteristics. A third approach is depicted as option C: drug release is induced from liposomal bilayers by external stimuli (local pH change or temperature change). Finally, one can envision that immunoliposomes can be built with intrinsic fuso-genic potential, which is only activated upon attach-ment of the carrier to the target cell. This exciting option D, Figure 35, resembles the behavior of certain viruses. Viruses offer interesting pathways as to how to enter target cells and how to deliver their payload successfully in a target organelle (i.e., for viruses the nucleus). This virus-mimicking approach led to the design of artificial viruses for targeted delivery of genetic material, but this can be extended to ther-apeutic proteins (Mastrobattista et al., 2006).
Related Topics