Chapter: Basic Concept of Biotechnology : Tools and Techniques in Biotechnology
DNA Sequencing Techniques - Techniques of Biotechnology and Innovations
DNA SEQUENCING TECHNIQUES
Sequencing means to find the order of nucleotides on a string of DNA. A variation in a DNA sequence can lead to a changed or non-functional protein, and lead to a genetic disorder. Sequencing of DNA is important to identify the type of mutations in genetic diseases.
The four best known DNA sequencing techniques are:
i) Sanger method and its most important variants (enzymic methods);
ii) Maxam & Gilbert method and other chemical methods;
iii) PyrosequencingTM method – DNA sequencing in real time by the detection of released pyrophosphate (PPi) ; and
iv) Single molecule sequencing with exonuclease (exonuclease digestion of a single molecule composed of a single strand of fluorescently labelled deoxynucleotides).
i) Sanger's method and other enzymatic methods
Initially known as the chain termination method or the dideoxynucleotide method, it is comprised of a catalysed enzymatic reaction that polymerizes the DNA fragments complementary to thetemplate DNA of interest i.e. unknown DNA. A 32P-labelled primer, short oligonucleotide with a sequence complementary to the template DNA, is annealed to a precise known region on the template DNA, which provides an initial point for DNA synthesis. Catalytic polymerization of deoxynucleoside triphosphates (dNTP) occurs onto the DNA in the presence of DNA polymerases. The polymerization is extended and the enzyme incorporates a modified nucleoside into the growing chain. Different approaches, shotgun (random) and primer walking (direct) sequencing mostly, are used and their strategies are described below in detail.
Random approach
In the usual procedure, with few exceptions, there is no control of the region that is going to be sequenced. It is also known as shotgun sequencing. It has ready availability of optimized cloning vectors, fluorescently labelled universal primers also and software for the purpose of base calling and sequence assembly. The whole process comprises high level of automation, from the cloning of the vectors and colony selection until the bases called.
Direct approach
In direct sequencing, the sequence is known of unknown DNA. This approach is also known as primer walking (Fig. 9). Its major advantage is the reduced redundancy because of the direct nature of the approach which is opposite of random approach. However, this approach requires the synthesis of each new primer, which is expensive.
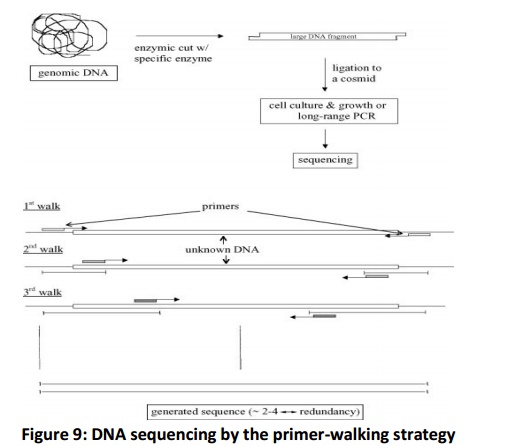
The genomic DNA is cut into a large piece (~ 40 kbp) and inserted into a cosmid for growth. The sequencing is performed by walks i.e. starting first from the known region of the cosmid. After the results from the first round are edited, a new priming site is located within the newly generated sequence. This procedure is repeated and gets over only when walks reach the opposite starting points.
Enzyme technology
Developments in DNA polymerase enzymes have significantly helped in the quality of the sequencing reactions and sequencing data. In fluorescently labelled dye-terminators, there was significant variation in peak intensity. The system of the termination was reproducible and predictable, but this deviation made automatic base calling difficult. Afew years later, a modified set of fluorescent labels for ddNTPs are introduced. The signal was more even, and automated base calling improved significantly.
Sample preparation
The following steps are often included in the methodology for sample preparation: (i) DNA scission and cloning into a vector (e.g. M13 or M13mp18); (ii) vector amplification to produce a phage-infected culture; and (iii) purification from the cell culture to yield pure single-stranded (ss) DNA template.
Labels and DNA labelling
A) Radioisotopes
The enzymic method used 32P as a label in the iitial stage. Biggin et al. (1983) suggested the use of deoxyadenosine 5’-(α-[35S] thio)triphosphate as the label unifiedinto the DNA fragments. This strategy stemmed in intensification of the band sharpness on autoradiography and the resolution of band separation.
B) Chemiluminescent detection
Chemiluminescent detection is used as an alternative to radioisotopes, a method based on Biotin – streptavidin system. The 5’-end of an oligonucleotide linked to biotin is used as the primer in the sequencing reaction. There are three major advantages to this method; first, the sequencing reactions are obtained directly from the PCR products; secondly, this method does not require cloning of the DNA before sequencing, and thirdly, it is feasible to multiplex several reactions on the same gel and detect one at a time with suitable enzyme linked primers.
C) Fluorescent dyes
Smith et al. (1986) developed a set of four different fluorescent dyes that allowed four reactions to be separated in a single lane.
Alternative dyes are synthesized. These dyes have emission spectra with their maxima relatively well spaced, which simplifies colour/base discrimination. One drawback of this group of dyes is the need for two wavelengths for excitation; for FAM and JOE dyes at 488 nm, and for TAMRA and ROX dyes at 543 nm.
ii) Maxam & Gilbert and other chemical methods
In this method, described by Maxam & Gilbert (1977), end-labelled DNA fragments are exposed to random cleavage at adenine, cytosine, guanine, or thymine positions by using specific chemical agents. The chemical attack is divided on three steps: base modification, removal of the modified base from its sugar, and DNA strand breaking at that sugar position. The products of these four reactions are then separated using polyacrylamide gel electrophoresis.
The chemical reactions can be separated into two different groups: (i) four-lane methods, where four (or more) separate cleavage procedures are used and the information is displayed in four (or more) parallel gel lanes and (ii) one-lane (or two-lane) method, where all reactions are based on only one chemical modification and electrophoresis is performed in a single (or two) lane(s).
iii) Pyrosequencing – DNA sequencing in real time by the detection of released PPi
Pyrosequencing, a real-time DNA-sequencing method, is based on the detection of the PPi released during the DNA polymerization reaction. Initially, this approach was used for continuous monitoring of DNApolymerase activity. Presently, there are two different pyro sequencing approaches: solid-phase sequencing and liquid-phase sequencing. The main problem noticed in all versions of pyrosequencing techniques is the interference of dATP in the detection of luminescence.
This problem is solved by replacing dATP by dATPαS in the experimental step.
Pyrosequencing has shown several advantages:
i) Detection is in real time with a cycle time of approximately 2 min (solid-phase) ;
ii) Reactions occur at room temperature and physiological pH;
iii) Method is easily adapted for multiplexed sample processing ;
On the other hand, this method has presented some disadvantages such as:
i) In the solid phase approach, the template must be washed completely after each nucleotide addition, resulting in a decreased signal due to loss of templates ;
ii) In the liquid phase approach, the apyrase activity is decreased in later cycles due to accumulation of intermediate products.
iii) Contamination with PPi decreases the signal-to-noise ratio significantly, due to increased background signal.
The main applications of this method are de novo DNA sequencing for short- and medium-length DNA, analysis of single-nucleotide polymorphisms, mutation detection and the analyses of secondary structure, such as hairpin structures.
Single-molecule sequencing with exonuclease
It was initially conceived as a laser-based technique allowing the fast sequencing of DNA fragments of 40 kb or more at a rate of 100 – 1000 bases/second. This technique is based on the identification of individual fluorescent nucleotides in a flowing sample stream. The method is divided into the following steps (Fig. 10): fluorescent labelling of the bases in a single fragment of DNA, attachment of this labelled DNA fragment onto a microsphere, movement of the supported DNA fragment into a flowing buffer stream, digestion of the labelled DNA with an exonuclease that sequentially cleaves the 3’-end nucleotides, anddetection and identification of individual fluorescently labelled bases as they cross a focused laser beam.
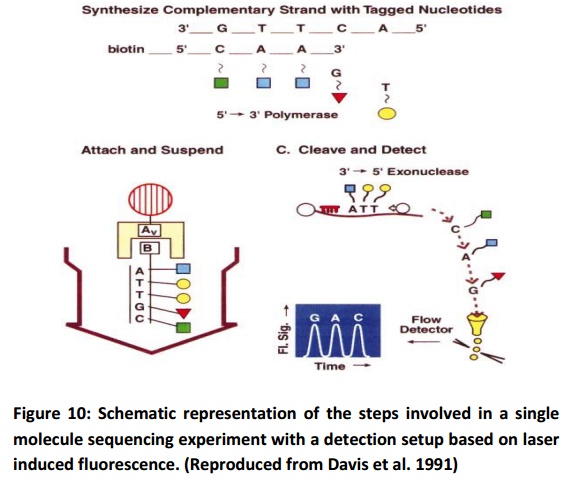
There are still many problems that have to be solved. The quality of buffer must be improved. A selection step must be incorporated into the sequencing process. Complementary DNA strands have to be labelled with four different nucleotides. New polymerases and new exonucleases are necessary for rapid and efficient sequencing.
Related Topics