Chapter: Modern Analytical Chemistry: Obtaining and Preparing Samples for Analysis
Classifying Separation Techniques: Separations Based on a Partitioning Between Phases
Separations Based on a Partitioning Between
Phases
The
most important class
of separation techniques is based on the selective parti- tioning of the analyte or interferent between
two immiscible phases.
When a phase containing a solute,
S, is brought into
contact with a second phase,
the solute parti- tions itself between the two phases.
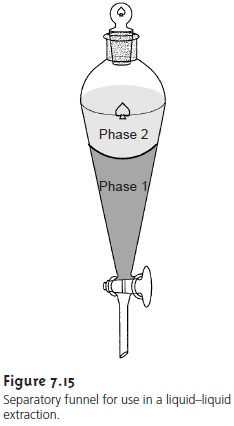
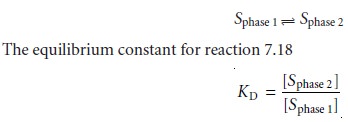 7.18
7.18
is
called the distribution constant, or partition coefficient. If KD is sufficiently large, then the solute will move from phase 1 to phase
2. The solute will remain
in phase 1, however, if the partition coefficient is sufficiently small. If a phase containing two solutes is brought
into contact with
a second phase,
and KD is favorable for only one of
the solutes, then
a separation of the solutes
may be possible. The physical states
of the two phases
are identified when describing the separation process,
with the phase containing the sample listed
first. For example,
when the sample
is in a liquid phase and the second phase
is a solid, the separation involves liquid–solid partitioning.
Extraction Between Two Phases
When the
sample is initially present in one
of the phases, the separation is known as an extraction. In a simple extraction the sample
is extracted one or more times with portions of the second
phase. Simple extrac- tions are particularly useful for separations in which only one component
has a fa- vorable distribution ratio. Several important separation
techniques are based on simple extractions, including
liquid–liquid, liquid–solid, solid–liquid, and gas–solid
extractions.
Liquid–Liquid Extractions
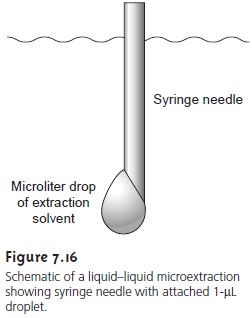
Liquid–liquid extractions are usually
accomplished with a separatory funnel
(Figure 7.15). The
two liquids are
placed in the
separa- tory funnel and shaken to increase the surface area between the phases. When the
extraction is complete, the liquids
are allowed to separate, with the denser
phase settling to the bottom of the separatory funnel. Liquid–liquid extractions also may be carried out
in the sample
container by adding
the extracting solvent
when the sample is collected. Pesticides in water, for example, may be preserved for longer periods
by extracting into
a small volume
of hexane added
to the sample
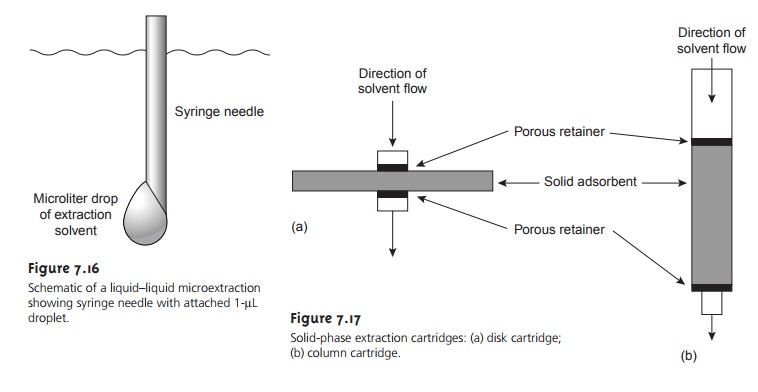
in the field. Liquid–liquid microextractions, in which
the extracting phase
is a 1-ÎĽL drop suspended from a microsyringe (Figure 7.16) also have been described.
Solid-Phase Extractions
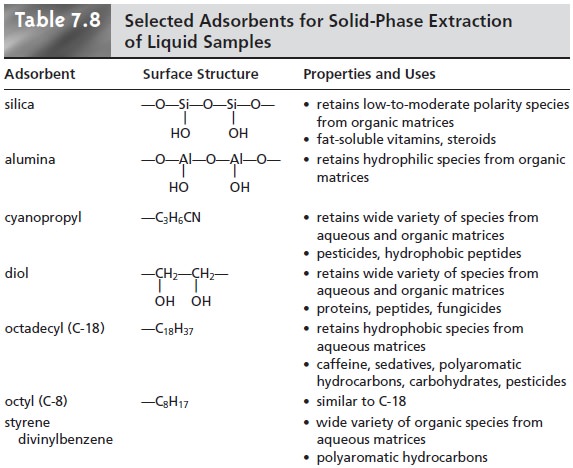
In a solid-phase extraction the sample is passed through a cartridge containing solid particulates that serve as the adsorbent material. For liq- uid samples the solid adsorbent is isolated in either a disk cartridge or a column (Figure 7.17). The choice of adsorbent is determined by the properties of the species being retained and the matrix in which it is found. Representative solid adsorbents are listed in Table 7.8.
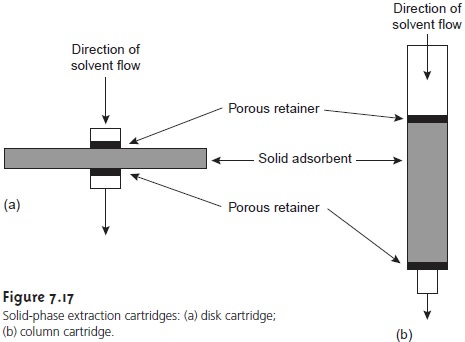
For example, sedatives, such as secobarbital and phenobarbi-
tal, can be isolated from
serum by a solid-phase extraction using a C-18
solid adsor- bent. Typically a 500-ÎĽL sample
of serum is passed through
the cartridge, with the
sedatives being retained
by a liquid–solid extraction. The cartridge is then washed with distilled water to remove any residual traces
of the serum’s matrix. Finally,
the retained sedatives are eluted from the cartridge by a solid–liquid
extraction using 500 ÎĽL of acetone.
For many analyses, solid–phase extractions are replacing
liquid–liquid extractions due to their
ease of use, faster extraction times, decreased
volumes of solvent, and their superior ability
to concentrate the analytes.
Solid-phase microextractions also
have been developed. In one approach, a fused silica fiber is placed inside a syringe needle.
The fiber, which is coated with a thin
organic film, such as poly(dimethyl siloxane), is lowered
into the sample
by de- pressing a plunger and exposed to the sample for a predetermined time. The fiber is
then
withdrawn into the needle and transferred to a gas chromatograph for analysis.
In gas–solid extractions the sample is passed through
a container packed
with a solid adsorbent. One example of the application of gas–solid extraction is in the analysis of organic compounds for carbon and hydrogen. The sample is combusted
in a flowing stream of O2, and
the gaseous combustion products are passed
through a series of solid-phase adsorbents that remove the CO2 and H2O.
Continuous Extractions
An extraction is still feasible even when the component of interest has an unfavorable partition coefficient, provided that all other components in the sample have significantly smaller partition coefficients.
Because the partition coefficient is unfavorable, a simple extraction will not be quantitative. Instead,
the extraction is accomplished by continuously passing
the extracting phase
through the sample until
a quantitative extraction is achieved.
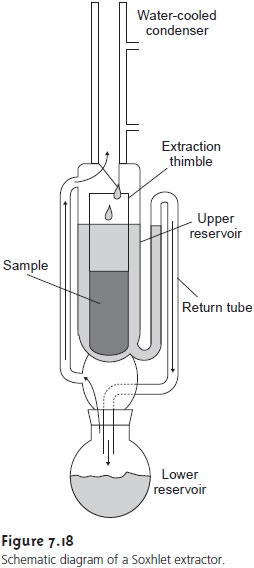
Many continuous extractions involving solid samples
are carried out with a Soxhlet extractor (Figure 7.18). The extracting solvent is placed in the lower reser- voir and heated to its boiling
point. Solvent in the vapor phase moves upward through the tube on the left side of the apparatus to the condenser where it con- denses back to the liquid state.
The solvent then passes through
the sample, which is
held in a porous cellulose filter thimble, collecting in the upper
reservoir. When the volume
of solvent in the upper
reservoir reaches the
upper bend of the return tube, the solvent and any extracted components are siphoned
back to the lower
reservoir. Over time,
the concentration of the extracted component in the lower
reservoir increases.
Soxhlet extractions have been replaced
in some applications by microwave-
assisted extractions.
The process is the same
as that described earlier for microwave digestion. The sample is placed in a sealed
digestion vessel along
with the liquid
ex- traction phase, and a microwave
oven is used to heat the extraction mixture. Using a sealed
digestion vessel allows
the extraction to take place
at a higher temperature
and pressure, thereby reducing the amount of time needed
for a quantitative extrac-
tion. In a Soxhlet extraction the temperature is limited by the solvent’s boiling point at atmospheric pressure. For example,
when acetone is the solvent,
a Soxhlet extrac- tion is limited to 56 °C. With a microwave-assisted extraction, however, a tempera- ture of over 150
°C can be obtained when
using acetone as the solvent.
Two
other examples of a continuous extraction deserve mention.
Volatile or- ganic compounds (VOCs) can be quantitatively removed
from liquid samples
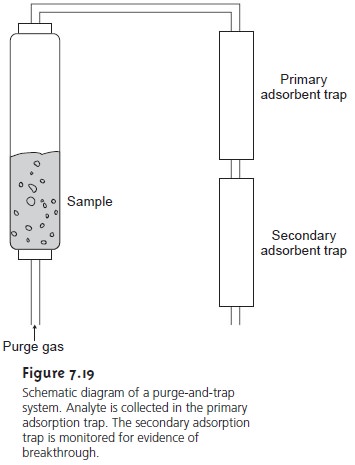
by a liquid–gas extraction. As shown in Figure 7.19,
the VOCs are removed by passing
an inert purging gas, such as He, through the sample. The He removes
the VOCs, which are then carried
by the He to a tube where
they are collected on a solid
adsor- bent. When the extraction is complete, the VOCs can then be removed from the
trap for analysis by rapidly
heating the tube while flushing
with He. This technique
is known as a purge and trap. Recoveries for analytes using
a purge and
trap may not be reproducible, requiring the use of internal standards for quantitative work.
Continuous extractions also can be accomplished with supercritical fluids.19 When a substance is heated above
its critical temperature and pressure, it forms a supercritical fluid whose properties are between those
of a gas and a liquid. Super- critical fluids are better solvents than gases, making them a better reagent
for ex- tractions. In addition, the viscosity of a supercritical fluid is significantly less than that of a liquid
solvent, allowing it to pass
more readily through
particulate samples. One example
of a supercritical extraction is the determination of total petroleum hydrocarbons (TPHs) in soils,
sediments, and sludges with supercritical CO2. Approximately 3 g of sample is placed in a 10-mL stainless steel cartridge, and super-
critical CO2, at a pressure
of 340 atm and a temperature of 80 °C, is passed
through the cartridge for
30 min at flow rate
of 1–2 mL/min.
The petroleum hydrocarbons are collected by passing
the effluent from the cartridge through 3 mL of tetra-
chloroethylene at room temperature. At this temperature the CO2 reverts
to the gas phase and is released to the atmosphere.
Chromatographic Separations
In
an extraction, the
sample is initially present in one phase,
and the component of interest is extracted into a second
phase. Separa- tions can
also be accomplished by continuously passing
one sample-free phase, called the mobile phase,
over a second sample-free phase
that remains fixed
or sta- tionary. The
sample is then
injected or placed
into the mobile
phase. As the
sam- ple’s components move with the mobile phase, they partition
themselves between the mobile and stationary phases. Those components having the largest
partition coefficients are more likely to move into the stationary phase, taking longer to pass through the system. This is the basis of all chromatographic separation techniques.
As currently practiced, modern chromatography provides a means
both of separat- ing analytes and interferents and of performing a qualitative or quantitative analysis of the analyte.
Related Topics