Chapter: Biology of Disease: Disorders of the Immune System
Antibody deficiencies - Primary Immunodeficiency Disease
Antibody deficiencies
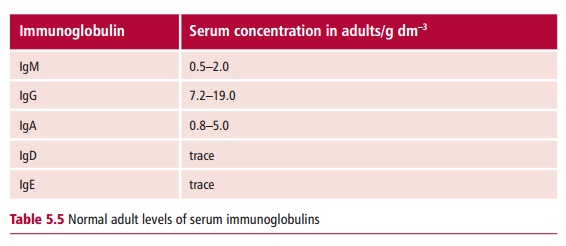
The normal concentration ranges for the five immunoglobulin classes, IgM, IgG, IgA, IgE and IgD , in adults are shown in Table 5.5. Deficiencies involving immunoglobulins of all classes are commonly referred to as agammaglobulinemias orhypogammaglobulinemias, depending on the level of deficiency. However, with some disorders there may be a selective deficiency of a single immunoglobulin class, as in selective IgA deficiency, or a dysregulation, where some antibody classes are reduced while others are increased.
During the first three months postpartum, the maternally derived IgG is catabolized and steadily disappears from the baby’s circulation. Between three and six months, serum IgG levels may be quite low, after which the levels begin to increase, and should normally attain ‘adult’ levels at about 12–18 months of age. In infants with transient hypogammaglobulinemia, the production of IgG is delayed considerably, sometimes for as long as two years. During this time the child is susceptible to recurrent infections withpyogenic (pus producing) bacteria and antibiotics must be administered. The incidence of TH has been estimated as 23 to 61 per million births.
Common variable immunodeficiency (CVID) is the commonest primary immunodeficiency involving all classes of antibody. It is a heterogeneous group of disorders and includes a range of phenotypes. Many patients are not diagnosed until early adulthood. Most patients show low levels of IgG and IgA, with near normal or 50% of normal levels of IgM and normal lymphocyte counts. The latter allows CVID to be distinguished from other antibody deficiencies such as X-linked agammaglobulinemia (see below). Some patients also have impaired cell-mediated immunity . The incidence has been estimated at one in 10 000–50 000.
The etiology of CVID is unknown and the majority of cases are sporadic. The B lymphocytes are immature and, when stimulated, do not differentiate into antibody-secreting plasma cells following the binding of an antigen, owing to defects in their cell surface receptors or signal transductionmechanisms . However, it is possible that in some patients there may be other defects, such as mutations in immunoglobulin regulatory genes. In addition, many CVID patients have defects in CD4+ T lymphocytes so that the T and B cell interactions required for antibody production are impaired.
Patients with CVID present with recurrent pyogenic infections, especially of the respiratory tract, commonly involving Streptococcus pneumoniae,Staphylococcus aureus, Haemophilus influenzae and Moraxella catarrhalis.
This can lead to bronchiectasis, in which the bronchi and bronchioles are abnormally dilated. Those patients who also have some impairment in cell-mediated immunity also suffer infections with mycobacteria and the fungus Pneumocystis carinii. Such patients also suffer recurrent severe infections withherpes simplex and herpes zoster , and may develop viral illness when immunized with live viral vaccines.
The diagnosis of CVID relies on the measurement of immunoglobulins, including specific antibodies to common vaccines, and ruling out other immunodeficiencies, such as X-linked agammaglobulinemia. Its treatment usually involves replacement therapy with pooled immunoglobulins obtained from healthy donors. All pooled immunoglobulin preparations are treated to inactivate viruses, such as hepatitis virus or HIV, which may be present. These preparations are available commercially and need to be given intravenously every three to four weeks to maintain plasma levels and protect against infections. The dose depends on the weight of the patient and is usually 400–600 mg kg–1. Alternatively, weekly subcutaneous administration of lower doses, which can be done at home, may be more convenient. Intramuscular injection, which allows a greater volume to be administered than can be delivered subcutaneously, can also be given.
X-linked agammaglobulinemia (XLA) or Bruton’s disease is caused by a deficiency in Bruton’s tyrosine kinase (Btk), which is required for the maturation of preB cells in the bone marrow to form B lymphocytes. The deficiency of Btk is due to one or more of 300 different mutations in the BTK gene located on the X chromosome. B lymphocytes are therefore absent from the circulation and plasma cells are not present in the spleen and lymph nodes. Tonsils and adenoids may be absent, as demonstrated by radiography. However, circulating T lymphocytes are normal. Serum immunoglobulin levels are extremely low and all classes of immunoglobulin are affected. This is a rare inherited disorder with an incidence of about one in 250 000 males. Patients present with recurrent pyogenic infections, which occur from around three to nine months of age, when maternally derived IgG is low. Infections encountered may result in pneumonia, otitis media, meningitis and diarrhea.
It is essential that patients with XLA are diagnosed as early as possible so that replacement immunoglobulin therapy can begin. Infections are treated with antibiotics. The prognosis for children diagnosed before the age of five years is good, with patients often surviving to middle age. Tests for mutations of the BTK gene are available, which allows for genetic counseling of affected femalesand prenatal diagnosis of fetal cells obtained by chorionic villus sampling or amniocentesis , with the possibility of a therapeutic abortion.
Selective IgA deficiency, as its name implies, affects only a single class of immunoglobulin. Many IgA deficient individuals are asymptomatic, with the condition only being detected during investigation of other disorders. In contrast, other patients with selective IgA deficiency suffer recurrent infections, typically ear infections, sinusitis and pneumonia. A high proportion of sufferers also develop autoantibodies that are directed against a variety of self antigens and approximately a third present with autoimmune diseases, such as systemic lupus erythematosus (SLE). It is not known which features determine the severity of the disease. Selective IgA deficiency is a relatively common disorder with an incidence of one in 500 to 700 Caucasians, although the frequency is much lower in other ethnic groups.
A patient who presents with a history of recurrent infection, chronic diarrhea, and autoimmune disease should be suspected of having a selective IgA deficiency. This can be confirmed by measuring serum immunoglobulin concentrations. Values of IgA below 0.07 mg dm–3, while other immunoglobulins are normal, would confirm the deficiency. Treatment of selective IgA deficiency normally involves using antibiotics to treat bacterial infections and replacement therapy is not usually necessary. If the disease presents with autoimmunity then anti-inflammatory drugs, such as corticosteroids, may be given. The prognosis is good, with patients living normal lifespans. However, approximately 10% of patients with a selective IgA deficiency also have a deficiency of the IgG2 subclass, which is usually produced in response to polysaccharide antigens. Patients with both defects suffer more severe bacterial infections, especially with encapsulated bacteria. Immunoglobulin replacement therapy may be appropriate in these cases.
Related Topics