Chapter: Introduction to Human Nutrition: The Vitamins
Vitamin E - Human Nutrition
Vitamin E
Although vitamin E was identified as a dietary essen-tial for animals in the 1920s, it was not until 1983 that it was clearly demonstrated to be a dietary essential for human beings. Unlike other vitamins, no unequiv-ocal physiological function for vitamin E has been defined; it acts as a lipid-soluble antioxidant in cell membranes, but many of its functions can be replaced by synthetic antioxidants. There is epidemiological evidence that high intakes of vitamin E are associated with lower incidence of cardiovascular disease, although in many intervention trials vitamin E supplements have been associated with increased all-cause mortality.
Vegetable oils are rich sources of vitamin E, but significant amounts are also found in nuts and seeds, most green leafy vegetables, and a variety of fish.
Vitamers and units of activity
Vitamin E is the generic descriptor for two families of compounds, the tocopherols and the tocotrienols (Figure 8.5). The different vitamers have different biological potency. The most active is α-tocopherol, and it is usual to express vitamin E intake in terms of mg α-tocopherol equivalents. This is the sum of mg α-tocopherol + 0.5 × mg β-tocopherol + 0.1 × mg γ-tocopherol + 0.3 × mg α-tocotrienol. The other vitamers have negligible vitamin activity.
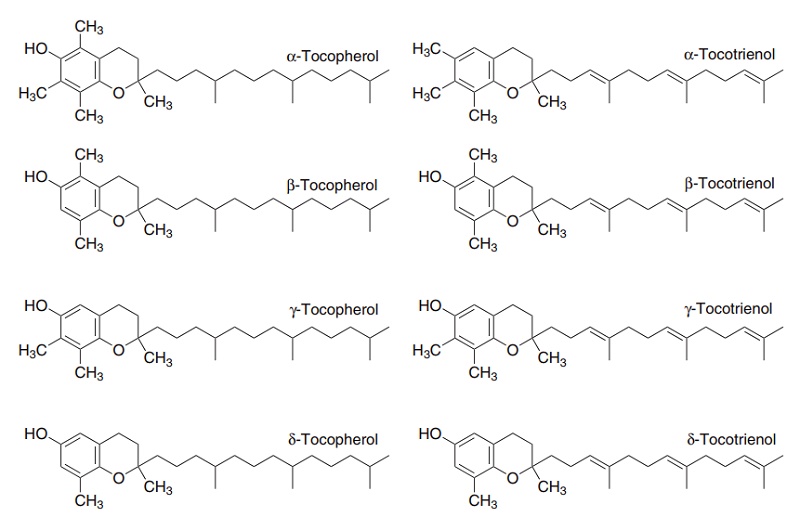
Figure 8.5 The vitamin E vitamers, tocopherols and tocotrienols.
The obsolete international unit of vitamin E activ-ity is still sometimes used: 1 IU = 0.67 mg α-tocopherol equivalent; 1 mg α-tocopherol = 1.49 IU.
Synthetic α-tocopherol does not have the same biological potency as the naturally occurring com-pound. This is because the side-chain of tocopherol has three centers of asymmetry and when it is synthe-sized chemically the result is a mixture of the various isomers. In the naturally occurring compound all three centers of asymmetry have the R-configuration, and naturally occurring α-tocopherol is called all-R, or RRR-α-tocopherol.
Absorption and metabolism
Tocopherols and tocotrienols are absorbed unchanged from the small intestine, in micelles with other dietary lipids, and incorporated into chylomicrons. The major route of excretion is in the bile, largely as gluc-uronides and other conjugates.
There are two mechanisms for tissue uptake of vitamin E. Lipoprotein lipase releases the vitamin by hydrolyzing the triacylglycerols in chylomicrons and VLDLs, while separately there is uptake of low-density lipoprotein (LDL)-bound vitamin E by means of LDL receptors. Retention within tissues depends on intra-cellular binding proteins, and it is likely that the dif-ferences in biological activity of the vitamers are due to differences in the affinity of these proteins for the different vitamers.
Metabolic functions of vitamin E
The main function of vitamin E is as a radical-trapping antioxidant in cell membranes and plasma lipoproteins. It is especially important in limiting radical damage resulting from oxidation of PUFAs, by reacting with the lipid peroxide radicals before they can establish a chain reaction. The tocopheroxyl radical formed from vitamin E is relatively unreactive and persists long enough to undergo reaction to yield non-radical products. Commonly, the vitamin E radical in a membrane or lipoprotein is reduced back to tocopherol by reaction with vitamin C in plasma. The resultant monodehydroascorbate radical then undergoes enzymic or non-enzymic reaction to yield ascorbate and dehydroascorbate, neither of which is a radical.
The stability of the tocopheroxyl radical means that it can penetrate further into cells, or deeper into plasma lipoproteins, and potentially propagate a chain reaction. Therefore, although it is regarded as an antioxidant, vitamin E may, like other antioxidants, also have pro-oxidant actions at high
This may explain why, although epi-demiological studies have shown a clear association between high blood concentrations of vitamin E and lower incidence of atherosclerosis, the results of inter-vention trials have generally been disappointing. In many trials there has been increased all-cause mortal-ity among those taking vitamin E and other antioxi-dant supplements.
The tocotrienols have lower vitamin activity than tocopherols, and indeed it is conventional to consider only γ-tocotrienol as a significant part of vitamin E intake. However, because of their unsaturated side-chain, the tocotrienols also have a hypocholesterol-emic action not shared by the tocopherols. They act to reduce the activity of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG CoA) reductase, the rate-limiting enzyme in the pathway for synthesis of cholesterol, by repressing synthesis of the enzyme.
Vitamin E deficiency
In experimental animals vitamin E deficiency results in a number of different conditions.
● Deficient female animals suffer the death and reab-sorption of the fetuses. This provided the basis of the original biological assay of vitamin E.
● In male animals deficiency results in testicular atrophy and degeneration of the germinal epithe-lium of the seminiferous tubules.
● Both skeletal and cardiac muscle are affected in deficient animals. This necrotizing myopathy is sometimes called nutritional muscular dystrophy – an unfortunate term, since there is no evidence that human muscular dystrophy is related to vitamin E deficiency.
● The integrity of blood vessel walls is affected, with leakage of blood plasma into subcutaneous tissues and accumulation under the skin of a green fluid: exudative diathesis.
● The nervous system is affected, with the develop-ment of central nervous system necrosis and axonal dystrophy. This is exacerbated by feeding diets rich in PUFAs.
Dietary deficiency of vitamin E in human beings is unknown, although patients with severe fat malab-sorption, cystic fibrosis, some forms of chronic liver disease or (very rare) congenital lack of plasma β-lipoprotein suffer deficiency because they are unable to absorb the vitamin or transport it around the body.
They suffer from severe damage to nerve and muscle membranes.
Premature infants are at risk of vitamin E defi-ciency, since they are often born with inadequate reserves of the vitamin. The red blood cell membranes of deficient infants are abnormally fragile, as a result of unchecked oxidative radical attack. This may lead to hemolytic anemia if they are not given supple-ments of the vitamin.
Experimental animals that are depleted of vitamin E become sterile. However, there is no evidence that vitamin E nutritional status is in any way associated with human fertility, and there is certainly no evi-dence that vitamin E supplements increase sexual potency, prowess, or vigor.
Vitamin E requirements
It is difficult to establish vitamin E requirements, partly because deficiency is more or less unknown, but also because the requirement depends on the intake of PUFAs. It is generally accepted, albeit with little experimental evidence, that an acceptable intake of vitamin E is 0.4 mg α-tocopherol equivalent/g dietary PUFA.
Indices of vitamin E status
The plasma concentration of α-tocopherol is used to assess vitamin E status; since most vitamin E is trans-ported in plasma lipoproteins, it is the concentration per gram total plasma lipid, or better per mole cholesterol, that is useful, rather than the simple concentration.
Erythrocytes are incapable of de novo lipid synthe-sis, so peroxidative damage resulting from oxygen stress has a serious effect, shortening red cell life and possibly precipitating hemolytic anemia in vitamin E deficiency. This has been exploited as a method of assessing status by measuring the hemolysis of red cells induced by dilute hydrogen peroxide relative to that observed on incubation in water. This gives a means of assessing the functional adequacy of vitamin E intake, albeit one that will be affected by other, unre-lated, factors. Plasma concentrations of α-tocopherol below 2.2 mmol/mol cholesterol or 1.1 μmol/g total plasma lipid are associated with increased susceptibil-ity of erythrocytes to induced hemolysis in vitro.
An alternative method of assessing functional anti-oxidant status, again one that is affected by both vitamin E and other antioxidants, is by measuring the exhalation of pentane arising from the catabolism of the products of peroxidation of n-6 PUFAs or ethane arising from n-3 PUFAs.
Higher levels of intake
There is good epidemiological evidence that intakes of vitamin E are associated with a lower risk of ath-erosclerosis and ischemic heart disease. High concen-trations of vitamin E will inhibit the oxidation of PUFAs in plasma lipoproteins, and it is this oxidation that is responsible for the development of atheroscle-rosis. The plasma concentrations of α-tocopherol that appear to be beneficial would require an intake of 17–40 mg/day, which is above what could be achieved by eating normal diets. Individual interven-tion trials of vitamin E supplements have generally been disappointing, and metaanalysis shows a signifi-cant increase in all-cause mortality among people taking vitamin E (and other antioxidant) supple-ments. This presumably reflects the fact that the stable tocopheroxyl radical can penetrate deeper into tissues and plasma lipoproteins, and increase radical damage. However, it is also possible that the plasma concentra-tion of α-tocopherol is a surrogate marker for some other protective factor in the diet.
Interactions with other nutrients
Vitamin C in plasma and extracellular fluid is impor-tant in reducing the tocopheroxyl radical in cell membranes and plasma lipoproteins back to tocoph-erol. There is also evidence that a variety of lipid-soluble antioxidants may be important in the antioxidant action of vitamin E in membranes and lipoproteins, including ubiquinone and synthetic antioxidants used in food processing, such as butyl-ated hydroxytoluene and butylated hydroxyanisole. Synthetic antioxidants will prevent or cure a number of the signs of vitamin E deficiency in experimental animals.
There is a considerable overlap between the func-tions of vitamin E and selenium. Vitamin E reduces lipid peroxide radicals to unreactive fatty acids; the selenium-dependent enzyme glutathione peroxidase reduces hydrogen peroxide to water, thus lowering the intracellular concentration of potentially lipid-dam-aging peroxide. A membrane-specific isoenzyme of glutathione peroxidase will also reduce the tocopher-oxyl radical back to tocopherol. Thus, vitamin E acts to remove the products of lipid peroxidation, whereas selenium acts both to remove the cause of lipid per-oxidation and to recycle vitamin E.
Related Topics