Chapter: Introduction to Human Nutrition: The Vitamins
Vitamin D - Human Nutrition
Vitamin D
Vitamin D is not strictly a vitamin, since it can be synthesized in the skin, and indeed under most conditions endogenous synthesis is the major source of the vitamin: it is only when sunlight exposure is inadequate that a dietary source is required. Its main func-tion is in the regulation of calcium absorption and homeostasis; most of its actions are mediated by nuclear receptors that regulate gene expression. Deficiency, leading to rickets in children and osteo-malacia in adults, continues to be a problem in north-ern latitudes, where sunlight exposure is poor.
There are relatively few sources of vitamin D, mainly oily fish, with eggs, liver, and butter providing modest amounts; fortified milk, containing ergocalciferol, is available in some countries. As a result, strict vegetar-ians are especially at risk of deficiency, especially in northern latitudes with little sunlight exposure.
Although meat provides apparently negligible quantities of vitamin D, it may be an important source, since what is present is largely the final active metabolite, calcitriol, which is many times more potent on a molar basis than is cholecalciferol.
Vitamers and international units
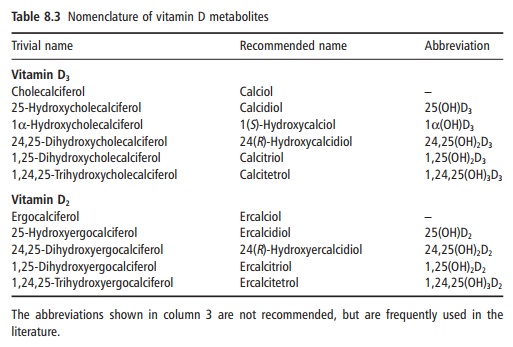
The normal dietary form of vitamin D is cholecalcif-erol (also known as calciol). This is also the com-pound that is formed in the skin by ultraviolet (UV) irradiation of 7-dehydrocholesterol. Some foods are enriched or fortified with (synthetic) ergocalciferol, which undergoes the same metabolism as cholecalcif-erol and has the same biological activity. Early studies assigned the name vitamin D1 to an impure mixture of products derived from the irradiation of ergosterol; when ergocalciferol was identified it was called vitamin D2, and when the physiological compound was identified as cholecalciferol it was called vita-min D3.
Like vitamin A, vitamin D was originally measured in international units of biological activity before the pure compound was isolated: 1 IU = 25 ng of chole-calciferol; 1 μg of cholecalciferol = 40 IU.
Absorption and metabolism
Vitamin D is absorbed in lipid micelles and incorpo-rated into chylomicrons; therefore, people on a low-fat diet will absorb little of such dietary vitamin D as is available. Indeed, it is noteworthy that at the time that rickets was a major public health problem in Scotland, herrings (a rich source) were a significant part of the diet: it can only be assumed that the diet was so low in fat that the absorption of the vitamin was impaired.
Synthesis of vitamin D in the skin
As shown in Figure 8.4, the steroid 7-dehydrocholes-terol (an intermediate in the synthesis of cholesterol that accumulates in the skin but not other tissues) undergoes a non-enzymic reaction on exposure to UV light, yielding previtamin D, which undergoes a further reaction over a period of hours to form cho-lecalciferol, which is absorbed into the bloodstream.
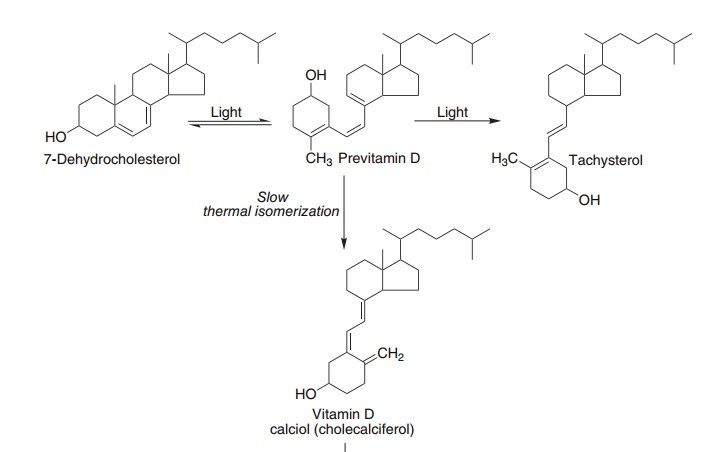
In temperate climates there is a marked seasonal variation in the plasma concentration of vitamin D; it is highest at the end of summer and lowest at the end of winter. Although there may be bright sunlight in winter, beyond about 40° N or S there is very little UV radiation of the appropriate wavelength for cho-lecalciferol synthesis when the sun is low in the sky. By contrast, in summer, when the sun is more or less overhead, there is a considerable amount of UV light even on a moderately cloudy day, and enough can penetrate thin clothes to result in significant forma-tion of vitamin D.
In northerly climates, and especially in polluted industrial cities with little sunlight, people may well not be exposed to enough UV light to meet their vitamin D needs, and they will be reliant on the few dietary sources of the vitamin.
Metabolism to calcitriol
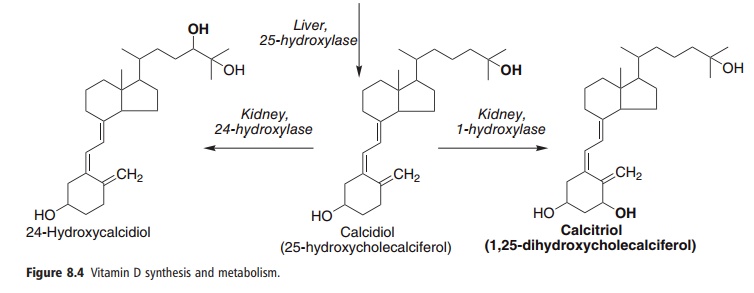
Cholecalciferol, either synthesized in the skin or from foods, undergoes two hydroxylations to yield the active metabolite, 1,25-dihydroxyvitamin D or cal-citriol, as shown in Figure 8.4. Ergocalciferol from fortified foods undergoes similar hydroxylation to yield ercalcitriol. The nomenclature of the vitamin D metabolites is shown in Table 8.3.
The first stage in vitamin D metabolism occurs in the liver, where it is hydroxylated to form the 25-hydroxy derivative calcidiol. This is released into the circulation bound to a vitamin D binding globulin. There is no tissue storage of vitamin D; plasma cal-cidiol is the main storage form of the vitamin, and it is plasma calcidiol that shows the most significant seasonal variation in temperate climates.
The second stage of vitamin D metabolism occurs in the kidney, where calcidiol undergoes either 1-hydroxylation to yield the active metabolite 1,25-dihydroxyvitamin D (calcitriol) or 24-hydroxylation to yield an apparently inactive metabolite, 24,25-dihydroxyvitamin D (24-hydroxycalcidiol). Calcidiol 1-hydroxylase is also found in other tissues that are capable of forming calcitriol as an autocrine or para-crine agent.
Regulation of vitamin D metabolism
The main function of vitamin D is in the control of calcium homeostasis and, in turn, vitamin D metabo-lism in the kidney is regulated, at the level of 1- or 24-hydroxylation, by factors that respond to plasma concentrations of calcium and phosphate. In tissues other than the kidney that hydroxylate calcidiol to calcitriol, the enzyme is not regulated in response to plasma calcium.
● Calcitriol acts to reduce its own synthesis and increase formation of 24-hydroxycalcidiol, by regu-lating the expression of the genes for the two hydroxylases.
● Parathyroid hormone is secreted in response to a fall in plasma calcium. In the kidney it acts to increase the activity of calcidiol 1-hydroxylase and decrease that of 24-hydroxylase. In turn, both cal-citriol and high concentrations of calcium repress the synthesis of parathyroid hormone; calcium also inhibits the secretion of the hormone from the parathyroid gland.
● Calcium exerts its main effect on the synthesis and secretion of parathyroid hormone. However, calcium ions also have a direct effect on the kidney, reducing the activity of calcidiol 1-hydroxylase.
● Phosphate also affects calcidiol metabolism; throughout the day there is an inverse fluctuation of plasma phosphate and calcitriol, and feeding people on a low-phosphate diet results in increased circulating concentrations of calcitriol.
Metabolic functions of vitamin D
The principal function of vitamin D is to maintain the plasma concentration of calcium; calcitriol achieves this in three ways:
● increased intestinal absorption of calcium
● reduced excretion of calcium by stimulating resorp-tion in the distal renal tubules (due to increased calbindin D synthesis)
● mobilization of bone mineral.
There is a growing body of evidence that low vitamin D status (but not such a degree of deficiency as to disturb calcium homeostasis) is associated with impaired glucose tolerance, insulin resistance and non-insulin dependent diabetes mellitus, as well as obesity and the low grade chronic inflammation asso-ciated with (especially abdominal) obesity. There is also evidence poor vitamin D status is a factor in the etiology of some cancers. Calcitriol has a variety of permissive or modulatory effects; it is a necessary, but not sufficient, factor, in:
● synthesis and secretion of insulin, parathyroid, and thyroid hormones;
● inhibition of production of interleukin by activated
T-lymphocytes and of immunoglobulin by acti-vated B-lymphocytes;
●differentiation of monocyte precursor cells;
●modulation of cell differentiation, proliferation and apoptosis.
In most of its actions, the role of calcitriol seems to be in the induction or maintenance of synthesis of calcium binding proteins, and the physiological effects are secondary to changes in intracellular calcium concentrations.
Calcitriol acts like a steroid hormone, binding to a nuclear receptor protein, commonly as a heterodimer with the RXR (vitamin A) receptor, then binding to hormone response elements on DNA and modifying the expression of one or more genes.
The best-studied actions of vitamin D are in the intestinal mucosa, where the intracellular calcium binding protein induced by vitamin D is essential for the absorption of calcium from the diet. Vitamin D also acts to increase the transport of calcium across the mucosal membrane by recruiting calcium trans-port proteins to the cell surface.
Calcitriol also raises plasma calcium by stimulating the mobilization of calcium from bone. It achieves this by activating osteoclast cells. However, it acts later to stimulate the laying down of new bone to replace the loss, by stimulating the differentiation and recruit-ment of osteoblasts.
Vitamin D deficiency: rickets and osteomalacia
Historically, rickets is a disease of toddlers, especially in northern industrial cities. Their bones are under-mineralized as a result of poor absorption of calcium in the absence of adequate amounts of calcitriol. When the child begins to walk, the long bones of the legs are deformed, leading to bow-legs or knock knees. More seriously, rickets can also lead to collapse of the ribcage and deformities of the bones of the pelvis. Similar problems may also occur in adolescents who are deficient in vitamin D during the adolescent growth spurt, when there is again a high demand for calcium for new bone formation.
Osteomalacia is the adult equivalent of rickets. It results from the demineralization of bone, rather than the failure to mineralize it in the first place, as is the case with rickets. Women who have little exposure to sunlight are especially at risk from osteomalacia after several pregnancies, because of the strain that preg-nancy places on their marginal reserve of calcium.
Osteomalacia also occurs in the older people. Here again the problem may be inadequate exposure to sunlight, but there is also evidence that the capacity to form 7-dehydrocholesterol in the skin decreases with advancing age, so that older people are more reliant on the few dietary sources of vitamin D.
Although vitamin D is essential for prevention and treatment of osteomalacia in older people, there is less evidence that it is beneficial in treating the other common degenerative bone disease of advancing age, osteoporosis, which is due to a loss of bone matrix, rather than enhanced release of calcium from bone with no effect on the organic matrix, as is seen in osteomalacia. The result is negative calcium balance and loss of bone mineral, but secondary to the loss of organic matrix, owing to progressive loss of estrogens and androgens, rather than failure of the vitamin D system.
Vitamin D requirements and reference intakes
It is difficult to determine requirements for dietary vitamin D, since the major source is synthesis in the skin. Before the development of methods for mea-surement of calcidiol the diagnosis of subclinical rickets was by detection of elevated alkaline phospha-tase in plasma; nowadays, the main criterion of ade-quacy is the plasma concentration of calcidiol.
In older people with little sunlight exposure, a dietary intake of 10 μg of vitamin D/day results in a plasma calcidiol concentration of 20 nmol/l, the lower end of the reference range for younger adults at the end of winter. Therefore, the reference intake for older people is 10 μg/day, whereas average intakes of vitamin D from unfortified foods are less than 4 μg/day.
There is increasing evidence that high vitamin D status is associated with a lower incidence of various cancers, diabetes, and the metabolic syndrome, sug-gesting that desirable intakes are higher than current reference intakes. Widespread fortification of foods would improve vitamin D status, but might also put a significant proportion of the population at risk of hypervitaminosis and hypercalcemia. Increased sun-light exposure will improve vitamin D status without the risks of toxicity, but excessive sunlight exposure is a cause of skin cancer. The main problem in trying to balance improved vitamin D status through increased sunlight exposure, and increased risk of skin cancer, is that there is very little information on the amount of sunlight exposure required for the synthesis of a given amount of vitamin D.
Vitamin D toxicity
During the 1950s, rickets was more or less totally eradicated in Britain and other temperate countries. This was due to enrichment of a large number of infant foods with vitamin D. However, a small number of infants suffered from vitamin D poisoning, the most serious effect of which is an elevated plasma concentration of calcium. This can lead to contrac-tion of blood vessels, and hence dangerously high blood pressure, and calcinosis, that is the calcification of soft tissues, including the kidney, heart, lungs, and blood vessel walls.
Some infants are sensitive to intakes of vitamin D as low as 50 μg/day. To avoid the serious problem of vitamin D poisoning in these susceptible infants, the extent to which infant foods are fortified with vitamin D has been reduced considerably. Unfortunately, this means that a small proportion, who have relatively high requirements, are now at risk of developing rickets. The problem is to identify those who have higher requirements and provide them with supplements.
The toxic threshold in adults is not known, but those patients suffering from vitamin D intoxication who have been investigated were taking supplements providing more than 250 μg/day.
Although excess dietary vitamin D is toxic, exces-sive exposure to sunlight does not lead to vitamin D poisoning. There is a limited capacity to form the precursor, 7-dehydrocholesterol, in the skin, and a limited capacity to take up cholecalciferol from the skin. Furthermore, prolonged exposure of previ-tamin D to UV light results in further reactions to yield lumisterol and other biologically inactive compounds.
Interactions with drugs and other nutrients
As discussed above, vitamin D receptors form het-erodimers with RXR, so that vitamin D-dependent functions require adequate, but not excessive, vitamin A status. A number of drugs, including barbiturates and other anticonvulsants, induce cytochrome P450, resulting in increased catabolism of calcidiol (and retinol), and cause drug-induced osteomalacia. The antituberculosis drug isoniazid inhibits cholecal-ciferol 25-hydroxylase in the liver, and prolonged administration can lead to the development of osteomalacia.
Strontium is a potent inhibitor of the kidney 1-hydroxylase, and strontium intoxication can lead to the development of vitamin D-resistant rickets or osteomalacia. Although there is normally little expo-sure to potentially toxic intakes of strontium, its salts are sometimes used to treat chronic lead intoxication.
Related Topics