Chapter: Introduction to Human Nutrition: The Vitamins
Vitamin A - Human Nutrition
Vitamin A
Vitamin A was the first vitamin to be discovered, ini-tially as an essential dietary factor for growth. It has a role in vision, as the prosthetic group of the light-sensitive proteins in the retina, and a major role in the regulation of gene expression and tissue differentia-tion. Deficiency is a major public health problem in large areas of the world, and prevention of vitamin A deficiency is one of the three micronutrient priorities of the World Health Organization (WHO) (the other two are iron and iodine).
Vitamers and international units
Two groups of compounds, shown in Figure 8.1, have vitamin A activity: retinol, retinaldehyde, and retinoic acid (preformed vitamin A); and a variety of caro-tenes and related compounds (collectively known as carotenoids) that can be cleaved oxidatively to yield retinaldehyde, and hence retinol and retinoic acid. Those carotenoids that can be cleaved to yield retinal-dehyde are known as provitamin A carotenoids.
Preformed vitamin A (mainly as retinyl esters) is found only in foods of animal origin. The richest source by far is liver, which may contain sufficient vitamin A to pose a potential problem for pregnant women, since retinol is teratogenic in excess. Caro-tenes are found in green, yellow, and red fruits and
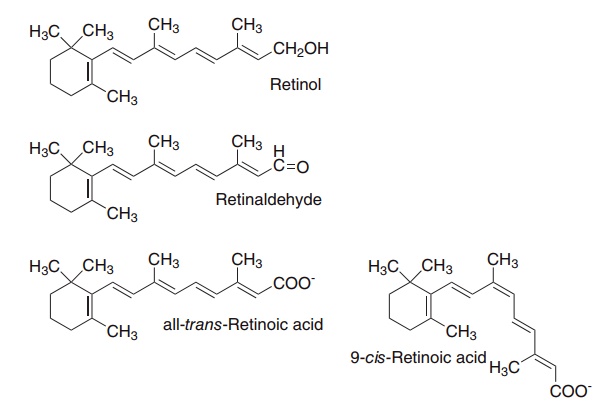
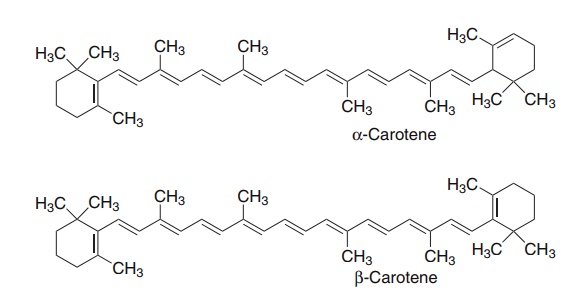
Figure 8.1 The major vitamin A vitamers and vitamin A active carotenoids
vegetables, as well as in liver, margarine, and milk and milk products. In addition to their role as precursors of vitamin A, carotenoids have potentially useful anti-oxidant action, and there is epidemiological evidence that diets that are rich in carotenoids (both those that are vitamin A active and those that are not) are associ-ated with a lower incidence of cancer and cardio-vascular disease. However, intervention studies with β-carotene have been disappointing, and it is not pos-sible to determine desirable intakes of carotene other than as a precursor of vitamin A.
Some 50 or more dietary carotenoids are potential sources of vitamin A: α-, β-, and γ-carotenes and cryptoxanthin are quantitatively the most important. Although it would appear from its structure that one molecule of β-carotene will yield two of retinol, this is not so in practice. Nutritionally, 6–12 μg of β-carotene is equivalent to 1 μg of preformed retinol. For other carotenes with vitamin A activity, 12–24 μg is equivalent to 1 μg of preformed retinol.
Conventionally, the total amount of vitamin A in foods is expressed as μg retinol equivalents, calculated from the sum of μg of preformed vitamin A +1/6 × μg β-carotene + 1/12 × μg other provitamin A carotenoids. Recent studies on the absorption of caro-tenes and their bioefficacy as vitamin A precursors have led to the definition of retinol activity equiva-lents. 1 μg retinol activity equivalent = 1 μg preformed retinol, 12 μg β-carotene or 24μg other provitamin A carotenoids.
Before pure vitamin A was available for chemical analysis, the vitamin A content of foods was deter-mined by biological assays and the results were expressed in standardized international units (IU): 1 IU = 0.3 μg of retinol, or 1 μg of retinol = 3.33 IU. Although obsolete, IU are sometimes still used in food labeling.
Metabolism and storage of vitamin A and pro-vitamin A carotenoids
Retinol is absorbed from the small intestine dissolved in lipid. About 70–90% of dietary retinol is normally absorbed, and even at high levels of intake this falls only slightly. However, in people with a very low fat intake (less than about 10% of energy from fat), absorption of both retinol and carotene is impaired, and low-fat diets are associated with vitamin A deficiency.
Dietary retinyl esters are hydrolyzed by lipases in the intestinal lumen and mucosal brush border mem-brane, then re-esterified to form retinyl palmitate before release into the circulation in chylomicrons.
Tissues can take up retinyl esters from chylomi-crons, but most retinol is in the chylomicron rem-nants that are taken up by the liver. Here retinyl esters are hydrolyzed, and the vitamin may either be secreted from the liver bound to retinol binding protein, or be transferred to stellate cells in the liver, where it is stored as retinyl esters in intracellular lipid droplets. Some 50–80% of the total body content of retinol is in the stellate cells of the liver, but a significant amount may also be stored in adipose tissue.
The main pathway for catabolism of retinol is oxidation to retinoic acid (which, as discussed below, has important biological activities in its own right, distinct from the activities of retinol). The main excretory product of both retinol and retinoic acid is retinoyl glucuronide, which is secreted in the bile.
As the intake of retinol increases, and the liver con-centration rises above 70 μmol/kg, a different pathway becomes increasingly important for the catabolism of retinol in the liver. This is a microsomal cytochrome P450-dependent oxidation, leading to a number of polar metabolites that are excreted in the urine and bile. At high intakes this pathway becomes saturated, and excess retinol is toxic since there is no further capacity for its catabolism and excretion.
Carotene dioxygenase
Like retinol, carotenoids are absorbed dissolved in lipid micelles. The biological availability and absorp-tion of dietary carotene varies between 5% and 60%, depending on the nature of the food, whether it is cooked or raw, and the amount of fat in the meal.
As shown in Figure 8.2, β-carotene and other pro-vitamin A carotenoids are cleaved in the intestinal mucosa by carotene dioxygenase, yielding retinalde-hyde, which is reduced to retinol, then esterified and secreted in chylomicrons together with retinyl esters formed from dietary retinol.
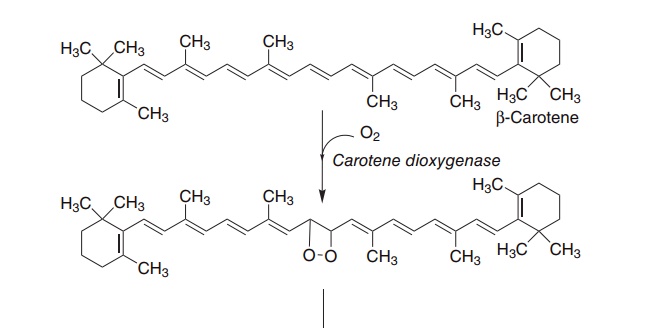
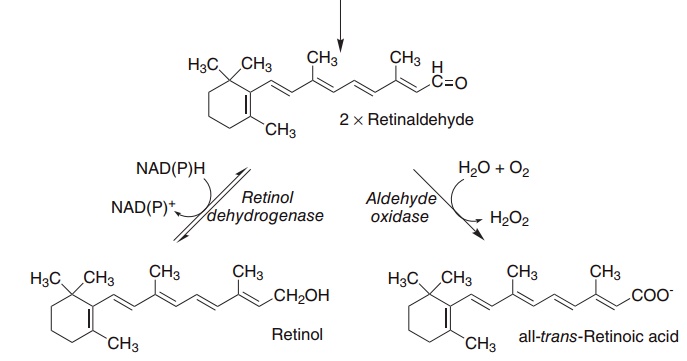
Figure 8.2 The oxidative cleavage of carotene to yield retinol and retinoic acid. Carotene dioxygenase (EC 1.13.11.21), retinol dehydrogenase (EC 1.1.1.105), retinaldehyde oxidase (EC 1.2.3.11).
Only a proportion of carotene undergoes oxidation in the intestinal mucosa, and a significant amount of carotene enters the circulation in chylomicrons. Caro-tene in the chylomicron remnants is cleared by the liver; some is cleaved by hepatic carotene dioxygenase, again giving rise to retinaldehyde and retinyl esters; the remainder is secreted in very low-density lipopro-teins (VLDLs), and may be taken up and cleaved by carotene dioxygenase in other tissues.
Central oxidative cleavage of β-carotene, as shown in Figure 8.2, should yield two molecules of retinal-dehyde, which can be reduced to retinol. However, as noted above, the biological activity of β-carotene, on a molar basis, is considerably lower than that of retinol, not twofold higher as might be expected. In addition to poor absorption of carotene, three factors may account for this.
●The intestinal activity of carotene dioxygenase is
relatively low, so that a relatively large proportion of ingested β-carotene may be absorbed unchanged.
●Other carotenoids in the diet may inhibit carotene dioxygenase and reduce the formation of retinol.
The principal site of carotene dioxygenase attack is the central bond of β-carotene, but asymmetric cleavage also occurs, leading to the formation of 8′-,10′- and 12′-apo-carotenals, which are oxidized to yield retinoic acid, but are not precursors of retinol or retinaldehyde.
Plasma retinol binding protein
Retinol is released from the liver bound to an α-globulin, retinol binding protein (RBP); this serves to maintain the vitamin in aqueous solution, protects it against oxidation and delivers the vitamin to target tissues. RBP is secreted from the liver as a 1:1 complex with the thyroxine-binding prealbumin, transthy-retin. This is important to prevent urinary loss of retinol bound to the relatively small RBP, which would otherwise be filtered by the kidney, with a consider-able loss of vitamin A from the body.
Cell surface receptors on target tissues take up retinol from the RBP–transthyretin complex, trans-ferring it on to an intracellular RBP. The receptors also remove the carboxy-terminal arginine residue from RBP, so inactivating it by reducing its affinity for both transthyretin and retinol. As a result, apo-RBP is filtered at the glomerulus; most is reabsorbed in the proximal renal tubules and hydrolyzed. The apopro-tein is not recycled.
During the development of vitamin A deficiency in experimental animals, the plasma concentration of RBP falls, whereas the liver content of apo-RBP rises. The administration of retinol results in release of holo-RBP from the liver. This provides the basis of the relative dose–response (RDR) test for liver reserves of vitamin A .
Metabolic functions of vitamin A and carotenes
The first function of vitamin A to be defined was in vision. More recently, retinoic acid has been shown to have a major function in regulation of gene expres-sion and tissue differentiation.
Vitamin A in vision
In the retina, retinaldehyde functions as the prosthetic group of the light-sensitive opsin proteins, forming rhodopsin (in rods) and iodopsin (in cones). Any one cone cell contains only one type of opsin, and hence is sensitive to only one color of light. Color blindness results from loss or mutation of one or other of the cone opsins.
In the pigment epithelium of the retina, all-trans-retinol is isomerized to 11-cis-retinol and then oxi-dized to 11-cis-retinaldehyde. This reacts with a lysine residue in opsin, forming the holoprotein rhodopsin. As shown in Figure 8.3, the absorption of light by rhodopsin causes isomerization of the retinaldehyde bound to opsin from 11-cis to all-trans, and a confor-mational change in opsin. This results in the release of retinaldehyde from the protein and the initiation of a nerve impulse. The overall process is known as bleach-ing, since it results in the loss of the color of rho-dopsin. The all-trans-retinaldehyde released from rhodopsin is reduced to all-trans-retinol, and joins the pool of retinol in the pigment epithelium for isomeri-zation to 11-cis-retinol and regeneration of rhodop-sin. The key to initiation of the visual cycle is the availability of 11-cis-retinaldehyde, and hence vitamin A. In deficiency both the time taken to adapt to dark-ness and the ability to see in poor light are impaired.
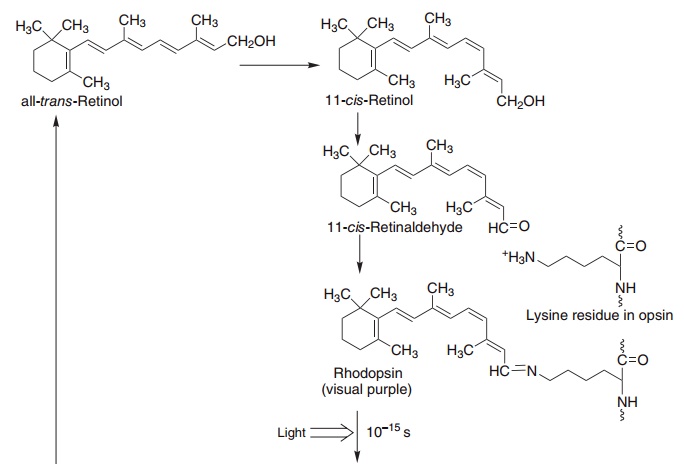
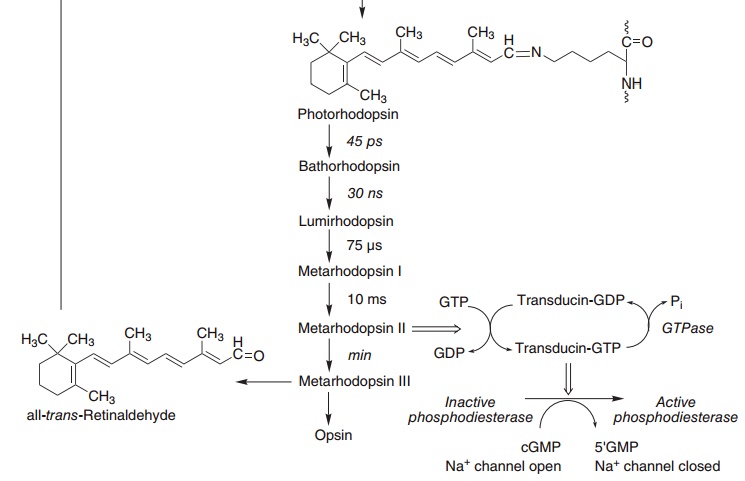
Figure 8.3 Role of vitamin A and the cyclic GMP cascade in the visual cycle. Retinol isomerase (EC 5.2.1.3), phosphodiesterase (EC 3.1.4.35).
The excited form of rhodopsin (metarhodopsin II) initiates a G-protein cascade leading to hyperpolar-ization of the outer section membrane of the rod or cone, caused by the closure of sodium channels through the membrane, and the initiation of a nerve impulse.
Retinoic acid and the regulation of gene expression
The main function of vitamin A is in the control of cell differentiation and turnover. All-trans-retinoic acid and 9-cis-retinoic acid are active in the regulation of growth, development, and tissue differentiation; they have different actions in different tissues. Like the steroid hormones and vitamin D, retinoic acid interacts with nuclear receptors that bind to response elements (control regions) of DNA, and regulate the transcription of specific genes.
There are two families of nuclear retinoid recep-tors: the retinoic acid receptors (RARs) bind all-trans-retinoic acid or 9-cis-retinoic acid, and the retinoid X receptors (RXRs) bind 9-cis-retinoic acid, and some of the other physiologically active retinoids as well. RXR can form active dimers with RARs, RXRs (homodimers), and the receptors for calcitriol (vitamin D), thyroid hormone, long-chain polyun-saturated fatty acid (PUFA) derivatives [the peroxi-some proliferators-activated receptor (PPAR)], and one for which the physiological ligand has not yet been identified (the COUP receptor).
The result of this is that a very large number of genes are sensitive to control by retinoic acid in different tissues, and at different stages in develop-ment, and retinoic acid is essential for the normal responses to vitamin D, thyroid hormone and long-chain PUFA derivatives.
Unoccupied RXRs can form dimers with calcitriol and other receptors; these bind to hormone response elements on DNA, but do not lead to activation of transcription. This means that vitamin A deficiency will impair responses to vitamin D and thyroid hormone more markedly than might be expected simply from lack of 9-cis-retinoic acid to form active heterodimers.
Vitamin A in excess may also impair responsiveness to vitamin D and other hormones, since high concen-trations of 9-cis-retinoic acid will lead to the forma-tion of RXR–RXR homodimers, leaving too few RXRs to form heterodimers with vitamin D and other receptors. There is epidemiological evidence that habitually high intakes of vitamin A are associated with poor bone health in later life as a result of impaired responsiveness to vitamin D.
The antioxidant function of carotenes
At least in vitro, and under conditions of low oxygen availability, carotenes can act as radical-trapping anti-oxidants. There is epidemiological evidence that high intakes of carotene are associated with a low incidence of cardiovascular disease and some forms of cancer, although the results of intervention trials with β-carotene have been disappointing, with an increased incidence of lung cancer among those taking carotene supplements.
The problem is that although carotene is an anti-oxidant at a low partial pressure of oxygen, as occurs in most tissues, at a high partial pressure of oxygen, as occurs in the lungs, it is an autocatalytic pro-oxidant, acting as a source of oxygen radicals. The UK Food Standards Agency specifically advises smokers not to take carotene supplements.
Vitamin A deficiency: night blindness and xerophthalmia
Worldwide, vitamin A deficiency is a major public health problem and the most important preventable cause of blindness; the WHO estimates that some 256 million children under 5 years old show subclinical deficiency and 2.7 million have xerophthalmia.
The earliest signs of clinical deficiency are associ-ated with vision. Initially, there is a loss of sensitivity to green light; this is followed by impairment of the ability to adapt to dim light, then an inability to see at all in dim light: night blindness. More prolonged or severe deficiency leads to the condition called xerophthalmia: keratinization of the cornea, followed by ulceration – irreversible damage to the eye that causes blindness. At the same time there are changes in the skin, with excessive formation of keratinized tissue.
Vitamin A also plays an important role in the dif-ferentiation of immune system cells, and mild defi-ciency, not severe enough to cause any disturbance of vision, leads to increased susceptibility to a variety of infectious diseases. At the same time, the synthesis of RBP is reduced in response to infection (it is a nega-tive acute-phase protein), so that there is a reduction in the circulating concentration of the vitamin, and hence further impairment of immune responses.
Signs of vitamin A deficiency also occur in protein– energy malnutrition, regardless of whether or not the intake of vitamin A is adequate. This is due to impair-ment of the synthesis of plasma RBP; functional vitamin A deficiency can occur secondary to protein– energy malnutrition; even if liver reserves of the vitamin are adequate, it cannot be mobilized.
Vitamin A requirements and reference intakes
There have been relatively few studies of vitamin A requirements in which subjects have been depleted of the vitamin for long enough to permit the develop-ment of clear deficiency signs. Current estimates of requirements are based on the intakes required to maintain a concentration in the liver of 70 μmol retinol/kg, as determined by measurement of the rate of metabolism of isotopically labeled vitamin A. This is adequate to maintain normal plasma concentra-tions of the vitamin, and people with this level of liver reserves can be maintained on a vitamin A-free diet for many months before they develop any detectable signs of deficiency.
The average requirement to maintain a concentra-tion of 70 μmol/kg of liver is 6.7 μg retinol equiva-lents/kg body weight, and this is the basis for calculation of reference intakes.
Assessment of vitamin A status
The only direct assessment of vitamin A status is by liver biopsy and measurement of retinyl ester reserves.
This is an invasive procedure that cannot be consid-ered for routine investigations and population surveys. Status can also be assessed by clinical and functional tests, the plasma concentrations of retinol and RBP, and the response to a test dose of vitamin A, the RDR test.
In field surveys, clinical signs of vitamin A defi-ciency, including Bitot’s spots, corneal xerosis, corneal ulceration, and keratomalacia, can be used to identify those suffering from vitamin A deficiency. The earliest signs of corneal damage are detected by conjunctival impression cytology (CIC); however, abnormalities only develop when liver reserves are seriously depleted.
The ability to adapt to dim light is impaired early in deficiency, and dark adaptation time is sometimes used to assess vitamin A status. However, the test is not suitable for use on children (the group most at risk of deficiency) and the apparatus is not suited to use in the field.
The fasting plasma concentration of retinol remains constant over a wide range of intakes and only falls significantly when liver reserves are nearly depleted. Therefore, although less sensitive to subtle changes within the normal range than some methods of assessing nutritional status, measurement of plasma retinol provides a convenient and sensitive means of detecting people whose intake of vitamin A is inade-quate to maintain normal liver reserves.
The RDR test is a test of the ability of a dose of retinol to raise the plasma concentration several hours after chylomicrons have been cleared from the circu-lation. It depends on the fact that apo-RBP accumu-lates in the liver in vitamin A deficiency. The RDR is the ratio of the plasma concentration of retinol 5 h after the dose to that immediately before it was given. An RDR greater than 20% indicates depletion of liver retinol to less than 70 μmol/kg.
Toxicity of vitamin A
There is only a limited capacity to metabolize vitamin A. Excessively high intakes lead to accumulation in the liver and other tissues, beyond the capacity of normal binding proteins, so that free, unbound, vitamin A is present. This leads to liver and bone damage, hair loss, vomiting, and headaches. Single doses of 60 mg of retinol are given to children in developing countries as a prophylactic against vitamin
A deficiency: an amount adequate to meet the child’s needs for 4–6 months. About 1% of children so treated show transient signs of toxicity, but this is considered an acceptable risk in view of the high prevalence and devastating effects of deficiency.
The chronic toxicity of vitamin A is a more general cause for concern; prolonged and regular intake of more than about 7.5–9 mg/day by adults (and signifi-cantly less for children) causes signs and symptoms of toxicity affecting:
●the central nervous system: headache, nausea, ataxia and anorexia, all associated with increased cerebro-spinal fluid pressure
●the liver: hepatomegaly with histological changes in the liver, increased collagen formation and hyperlipidemia
●bones: joint pains, thickening of the long bones, hypercalcemia and calcification of soft tissues the skin: excessive dryness, scaling and chapping of the skin, desquamation and alopecia.
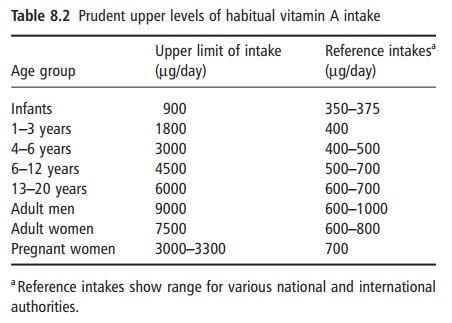
The recommended upper limits of habitual intake of retinol, compared with reference intakes, are shown in Table 8.2. As discussed above, habitual high intakes of vitamin A, albeit below these prudent upper levels of intake, may be associated with impaired respon-siveness to vitamin D, poor mineralization of bone and the early development of osteoporosis.
Teratogenicity of vitamin A
The synthetic retinoids (vitamin A analogues) used in dermatology are highly teratogenic. After women have been treated with them, it is recommended that contraceptive precautions be continued for 12 months, because of their retention in the body.
By extrapolation, it has been assumed that retinol is also teratogenic, although there is little evidence. In case– control studies, intakes between 2400μg/day and 3300 μg/day during pregnancy have been associated with birth defects. Other studies have not demon-strated any teratogenic effect at this level of intake, and it has been suggested that the threshold plasma concentration associated with teratogenic effects is unlikely to be reached with intakes below 7500 μg/ day. Nevertheless, pregnant women are advised not to consume more than 3000 μg/day (American Pediatric Association recommendation) or 3300 μg (UK Department of Health recommendation).
Interactions of vitamin A with drugs and other nutrients
Historically, there was considerable confusion between vitamins A and D, and for many years it was not clear which acted in which system. By the 1950s it was believed that the problem had been solved, with clearly defined functions of vitamin A in vision, and vitamin D in calcium homeostasis and bone develop-ment. However, both have overlapping effects on a number of systems, including bone metabolism and immune system function. It is now known that this is the result of formation of retinoid–vitamin D recep-tor heterodimers, so that in some systems both are required in appropriate amounts for normal regula-tion of gene expression.
Chlorinated hydrocarbons, as contained in agricul-tural pesticides, deplete liver retinol. Metabolites of polychlorinated biphenyls bind to the thyroxine binding site of transthyretin, and in doing so impair the binding of RBP. As a result there is free RBP-bound retinol in plasma, which is filtered at the glom-erulus and hence lost in the urine. Habitual use of barbiturates may also lead to deficiency as a result of induction of cytochrome P450, which catalyzes the catabolism of retinol.
Related Topics