Chapter: Introduction to Human Nutrition: The Vitamins
Vitamin B6 - Human Nutrition
Vitamin B6
Apart from a single outbreak in the 1950s, due to overheated infant milk formula, vitamin B6 deficiency is unknown except under experimental conditions. Nevertheless, there is a considerable body of evidence that marginal status and biochemical deficiency may be relatively widespread in developed countries.
Vitamin B6 is widely distributed in a variety of foods. However, a considerable proportion of the vitamin in plant foods may be present as glucosides, which are probably not biologically available, although a proportion may be hydrolyzed by intestinal bacteria.
When foods are heated, pyridoxal and pyridoxal phosphate can react with the ε-amino groups of lysine to form a Schiff base (aldimine). This renders both the vitamin B6and the lysine biologically unavailable; more importantly, the pyridoxyl-lysine released during digestion is absorbed and has antivitamin B6 antimetabolic activity. Overall, it is estimated that some 70–80% of dietary vitamin B6 is available.
Vitamers
The generic descriptor vitamin B6 includes six vita-mers: the alcohol pyridoxine, the aldehyde pyridoxal, the amine pyridoxamine, and their 5′-phosphates. There is some confusion in the older literature, because at one time “pyridoxine,” which is now used specifically for the alcohol, was used as a generic descriptor, with “pyridoxol” as the specific name for the alcohol. The vitamers are metabolically intercon-vertible and, as far as is known, they have equal bio-logical activity; they are all converted in the body to the metabolically active form, pyridoxal phosphate. 4-Pyridoxic acid is a biologically inactive end-product of vitamin B6 metabolism.
Absorption and metabolism
The phosphorylated vitamers are dephosphorylated by membrane-bound alkaline phosphatase in the intestinal mucosa; pyridoxal, pyridoxamine, and pyri-doxine are all absorbed rapidly by passive diffusion. Intestinal mucosal cells have pyridoxine kinase and pyridoxine phosphate oxidase (Figure 8.11), so that there is net accumulation of pyridoxal phosphate by metabolic trapping. Much of the ingested pyridoxine is released into the portal circulation as pyridoxal, after dephosphorylation at the serosal surface. Unlike other B vitamins, there seems to be no limit on the amount of vitamin B6 that is absorbed.
Most of the absorbed vitamin is taken up by the liver by passive diffusion, followed by metabolic trap-ping as phosphate esters, which do not cross cell membranes, then oxidation to pyridoxal phosphate. The liver exports both pyridoxal phosphate (bound to albumin) and pyridoxal (which binds to both albumin and hemoglobin). Free pyridoxal remaining in the liver is rapidly oxidized to 4-pyridoxic acid, which is the main excretory product.
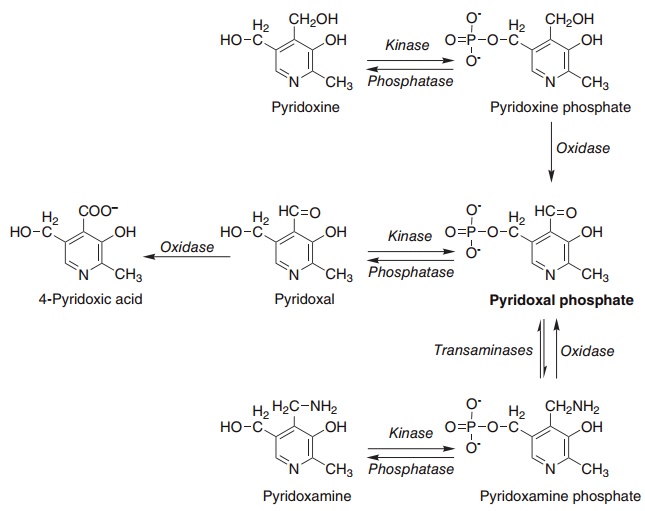
Figure 8.11 Interconversion of the vitamin B6 vitamers. Pyridoxal kinase (EC 2.7.1.38), pyridoxine phosphate oxidase (EC 1.1.1.65), pyridoxamine phosphate oxidase (EC 1.4.3.5).
Extrahepatic tissues take up pyridoxal and pyri-doxal phosphate from the plasma. The phosphate is hydrolyzed to pyridoxal, which can cross cell mem-branes, by extracellular alkaline phosphatase, then trapped intracellularly by phosphorylation. Tissue concentrations of pyridoxal phosphate are controlled by the balance between phosphorylation and dephosphorylation.
Some 80% of the body’s total vitamin B6 is pyri-doxal phosphate in muscle, mostly associated with glycogen phosphorylase. This does not function as a reserve of the vitamin and is not released from muscle in times of deficiency; it is released into the circula-tion (as pyridoxal) in starvation, when glycogen reserves are exhausted and there is less requirement for phosphorylase activity. Under these conditions it is available for redistribution to other tissues, and especially the liver and kidneys, to meet the increased need for transamination of amino acids to provide substrates for gluconeogenesis.
Metabolic functions of vitamin B6
Pyridoxal phosphate is a coenzyme in three main areas of metabolism:
● in a wide variety of reactions of amino acids, espe-cially transamination, in which it functions as the intermediate carrier of the amino group, and decar-boxylation to form amines
● as the cofactor of glycogen phosphorylase in muscle and liver, where it is the phosphate group that is catalytically important
● in the regulation of the action of steroid hormones. Pyridoxal phosphate acts to remove the hormone–
receptor complex from DNA binding, and so ter-minate the action of the hormones. In vitamin B6 deficiency there is increased sensitivity and respon-siveness of target tissues to low concentrations of steroid hormones, including estrogens, androgens, cortisol, and vitamin D.
Vitamin B6 deficiency
Deficiency of vitamin B6 severe enough to lead to clinical signs is extremely rare, and unequivocal defi-ciency has only been reported in one outbreak, during the 1950s, when babies were fed on a milk preparation that had been severely overheated during manufac-ture. Many of the affected infants suffered convul-sions, which ceased rapidly following the administra-tion of vitamin B6.
The cause of the convulsions was severe impair-ment of the activity of the pyridoxal phosphate-dependent enzyme glutamate decarboxylase, which catalyzes the synthesis of the inhibitory neurotrans-mitter γ-aminobutyric acid (GABA), together with accumulation of hydroxykynurenine as a result of impaired activity of kynureninase, which is also pyri-doxal phosphate dependent.
Moderate vitamin B6 deficiency results in a number of abnormalities of amino acid metabolism, espe-cially of tryptophan and methionine. In experimental animals, a moderate degree of deficiency leads to increased sensitivity of target tissues to steroid hormone action. This may be important in the devel-opment of hormone-dependent cancer of the breast, uterus, and prostate, and may therefore affect the prognosis. Vitamin B6 supplementation may be a useful adjunct to other therapy in these common cancers; certainly, there is evidence that poor vitamin B6 nutritional status is associated with a poor prog-nosis in women with breast cancer.
Vitamin B6 requirements
Most studies of vitamin B6 requirements have fol-lowed the development of abnormalities of trypto-phan and methionine metabolism during depletion and normalization during repletion with graded intakes of the vitamin. Although the tryptophan load test is unreliable as an index of vitamin B6 nutritional status in field studies, under the controlled conditions of depletion/repletion studies it gives a useful indica-tion of the state of vitamin B6 nutrition.
Since the major role of vitamin B6 is in amino acid metabolism it is likely that protein intake will affect vitamin B6 requirements. Adults maintained on vitamin B6-deficient diets develop abnormalities of tryptophan and methionine metabolism more quickly, and their blood vitamin B6 falls more rapidly, when their protein intake is relatively high (80–160 g/day in various studies) than on low protein intakes (30–50 g/ day). Similarly, during repletion of deficient subjects, tryptophan and methionine metabolism and blood vitamin B6 are normalized more rapidly at low than at high levels of protein intake.
From such studies the average requirement for vitamin B6 is estimated to be 13 μg/g dietary protein, and reference intakes are based on 15–16 μg/g dietary protein.
Estimation of the vitamin B6 requirements of infants presents a problem, and there is a clear need for further research. Human milk, which must be assumed to be adequate for infant nutrition, provides only some 2.5–3 μg of vitamin B6/g protein. This is very much lower than the requirement for adults, although there is no reason why infants should have a lower requirement.
Based on the body content of 3.7 μg (15 nmol) of vitamin B6/g body weight, and the rate of weight gain, a minimum requirement for infants over the first 6 months of life is 100 μg/day to establish tissue reserves, and an additional 20% to allow for metabolic turnover. Even if the mother receives daily supplements of 2.5 mg of vitamin B6through-out lactation, thus more than doubling her normal intake, the infant’s intake ranges from 100 μg/day to 300 μg/day over the first 6 months of life. At 1 month this is only 8.5 μg/g protein, rising to 15 μg/g by 2 months.
Assessment of vitamin B6 status
Fasting plasma total vitamin B6 (measured microbio-logically), or more specifically pyridoxal phosphate, is widely used as an index of vitamin B6 nutritional status. Despite the fall in plasma pyridoxal phosphate in pregnancy, which has been widely interpreted as indicating vitamin B6 depletion or an increased requirement, the plasma concentration of pyridoxal phosphate plus pyridoxal is unchanged. This suggests that determination of plasma pyridoxal phosphate alone may not be a reliable index of vitamin B6 nutri-tional status.
About half of the normal dietary intake of vitamin B6 is excreted as 4-pyridoxic acid. Urinary excretion of 4-pyridoxic acid will largely reflect the recent intake of the vitamin rather than the underlying nutritional status.
Coenzyme saturation of transaminases
The most widely used method of assessing vitamin B6 status is by the activation of erythrocyte transami-nases by pyridoxal phosphate added in vitro. An acti-vation coefficient for alanine transaminase >1.25, or for aspartate transaminase >1.8, is considered to indi-cate deficiency.
The tryptophan load test
The tryptophan load test for vitamin B6 nutritional status (the ability to metabolize a test dose of trypto-phan) is one of the oldest metabolic tests for func-tional vitamin nutritional status. It was developed as a result of observation of the excretion of an abnor-mal colored compound, later identified as the trypto-phan metabolite xanthurenic acid, in the urine of deficient animals.
Kynureninase (see Figure 8.12) is a pyridoxal phos-phate-dependent enzyme, and its activity falls mark-edly in vitamin B6 deficiency, at least partly because it undergoes a slow mechanism-dependent inactivation that leaves catalytically inactive pyridoxamine phos-phate at the active site of the enzyme. The enzyme can only be reactivated if there is an adequate supply of pyridoxal phosphate. This means that in vitamin B6 deficiency there is a considerable accumulation of both hydroxykynurenine and kynurenine, sufficient to permit greater metabolic flux than usual through kynurenine transaminase, resulting in increased for-mation of kynurenic and xanthurenic acids.
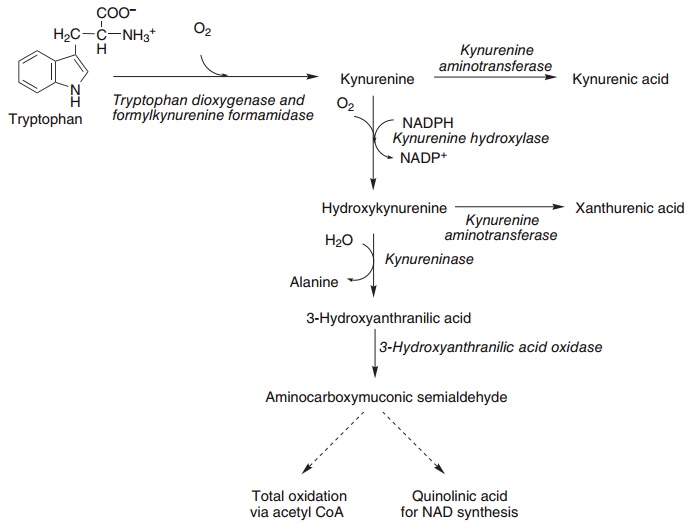
Figure 8.12 Oxidative pathway of tryptophan: the basis of the tryptophan load test. Tryptophan dioxygenase (EC 1.13.11.11), formylkynurenine formamidase (EC 3.5.1.9), kynurenine hydroxylase (EC 1.14.13.9), kynureninase (EC 3.7.1.3).
Xanthurenic and kynurenic acids, and kynurenine and hydroxykynurenine, are easy to measure in urine, so the tryptophan load test [the ability to metabolize a test dose of 2–5 g (150–380 μmol/kg body weight) of tryptophan] has been widely adopted as a con-venient and very sensitive index of vitamin B6 nutritional status. However, because glucocorticoid hormones increase tryptophan dioxygenase activity, abnormal results of the tryptophan load test must be regarded with caution, and cannot necessarily be interpreted as indicating vitamin B6 deficiency. Increased entry of tryptophan into the pathway will overwhelm the capacity of kynureninase, leading to increased formation of xanthurenic and kynurenic acids. Similarly, estrogen metabolites inhibit kyn-ureninase, leading to results that have been misinter-preted as vitamin B6 deficiency.
The methionine load test
The metabolism of methionine includes two pyri-doxal phosphate-dependent steps: cystathionine synthetase and cystathionase (see Figure 8.16). Cystathionase activity falls markedly in vitamin B6 deficiency, and as a result there is an increase in the urinary excretion of homocysteine and cystathionine, both after a loading dose of methionine and under
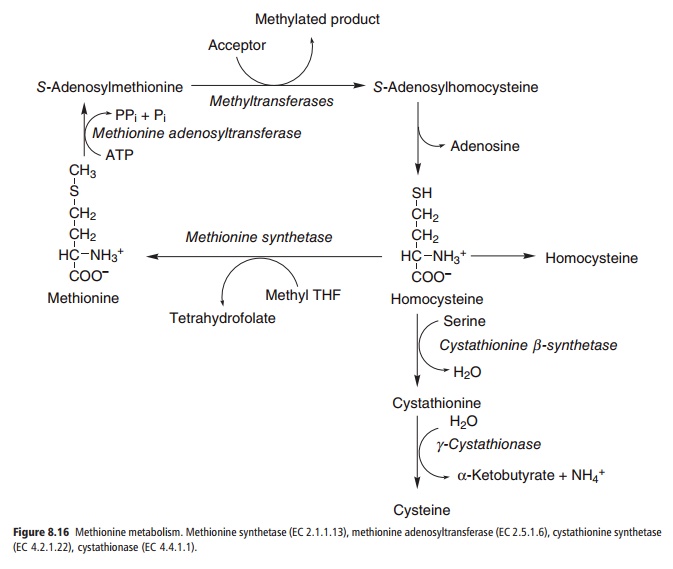
basal conditions. However, as discussed below, homo-cysteine metabolism is more affected by folate status than by vitamin B6 status and, like the tryptophan load test, the methionine load test is probably not reliable as an index of vitamin B6 status in field studies.
Non-nutritional uses of vitamin B6
Several studies have suggested that oral contraceptives cause vitamin B6 deficiency. As a result of this, supple-ments of vitamin B6 of 50–100 mg/day, and some-times higher, have been used to overcome the side-effects of oral contraceptives. Similar supplements have also been recommended for the treatment of the premenstrual syndrome, although there is little evi-dence of efficacy from placebo-controlled trials.
All of the studies that suggested that oral contra-ceptives cause vitamin B6 deficiency used the metabo-lism of tryptophan as a means of assessing vitamin B6 nutritional status. When other biochemical markers of status were also assessed, they were not affected by oral contraceptive use. Furthermore, most of these studies were performed using the now obsolete high-dose contraceptive pills.
Oral contraceptives do not cause vitamin B6 defi-ciency. The problem is that estrogen metabolites inhibit kynureninase and reduce the activity of kyn-urenine hydroxylase. This results in the excretion of abnormal amounts of tryptophan metabolites, similar to what is seen in vitamin B6 deficiency, but for a dif-ferent reason.
Doses of 50–200 mg of vitamin B6/day have an antiemetic effect, and the vitamin is widely used, alone or in conjunction with other antiemetics, to minimize the nausea associated with radiotherapy and to treat pregnancy sickness. There is no evidence that vitamin B6 has any beneficial effect in pregnancy sickness, or that women who suffer from morning sickness have lower vitamin B6 nutritional status than other pregnant women.
Doses of vitamin B6 of 100 mg/day have been reported to be beneficial in the treatment of the carpal tunnel syndrome or tenosynovitis. However, most of the reports originate from one centre and there appears to be little independent confirmation of the usefulness of the vitamin in this condition.
Vitamin B6 toxicity
In experimental animals, doses of vitamin B6 of 50 mg/kg body weight cause histological damage to dorsal nerve roots, and doses of 200 mg/kg body weight lead to the development of signs of peripheral neuropathy, with ataxia, muscle weakness, and loss of balance. The clinical signs of vitamin B6 toxicity in animals regress within 3 months after withdrawal of these massive doses, but sensory nerve conduction velocity, which decreases during the development of the neuropathy, does not recover fully.
Sensory neuropathy has been reported in seven patients taking 2–7 g of pyridoxine/day. Although there was some residual damage, withdrawal of these extremely high doses resulted in a considerable recovery of sensory nerve function. Other reports have suggested that intakes as low as 50 mg/day are associated with neurological damage, although these studies were based on patients reporting symp-toms rather than objective neurological examination. There have been no reports of nerve damage in chil-dren with vitamin B6-dependent homocystinuria, or other inborn errors of metabolism, who take 200–300 mg/day.
Related Topics