Chapter: Introduction to Human Nutrition: The Vitamins
Vitamin B2 (riboflavin) - Human Nutrition
Vitamin B2 (riboflavin)
Riboflavin deficiency is a significant public health problem in many areas of the world. The vitamin has a central role as a coenzyme in energy-yielding metabolism, yet deficiency is rarely, if ever, fatal, since there is very efficient conservation and recycling of riboflavin in deficiency.
The structures of riboflavin and the riboflavin-derived coenzymes are shown in Figure 8.9.
Milk and dairy products are important sources, providing 25% or more of total riboflavin intake in most diets, and it is noteworthy that average ribofla-vin status in different countries reflects milk con-sumption to a considerable extent. Other rich sources are eggs, meat, and fish. In addition, because of its intense yellow color, riboflavin is widely used as a food color.
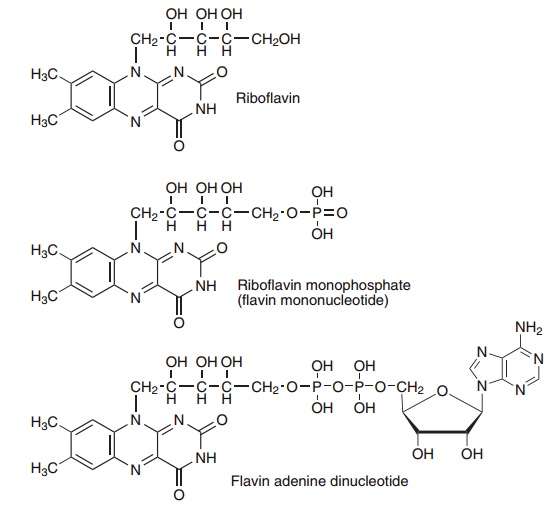
Figure 8.9 Riboflavin (vitamin B2) and the flavin coenzymes, riboflavin monophosphate and flavin adenine dinucleotide.
Photolytic destruction
Photolysis of riboflavin leads to the formation of lumiflavin (in alkaline solution) and lumichrome (in acidic or neutral solution), both of which are biologi-cally inactive. Exposure of milk in clear glass bottles to sunlight or fluorescent light can result in the loss of significant amounts of riboflavin. This is poten-tially nutritionally important. Lumiflavin and lumi-chrome catalyze oxidation of lipids (to lipid perox-ides) and methionine (to methional), resulting in the development of an unpleasant flavor, known as the “sunlight” flavor.
Absorption and metabolism
Apart from milk and eggs, which contain relatively large amounts of free riboflavin bound to specific binding proteins, most of the vitamin in foods is as flavin coenzymes bound to enzymes, which are released when the protein is hydrolyzed. Intestinal phosphatases then hydrolyze the coenzymes to liber-ate riboflavin, which is absorbed in the upper small intestine. The absorption of riboflavin is limited and after moderately high doses only a small proportion is absorbed.
Much of the absorbed riboflavin is phosphorylated in the intestinal mucosa and enters the bloodstream as riboflavin phosphate, although this does not seem to be essential for absorption of the vitamin.
About 50% of plasma riboflavin is free riboflavin, which is the main transport form, with 44% as flavin adenine dinucleotide (FAD) and the remainder as riboflavin phosphate. The vitamin is largely protein-bound in plasma; free riboflavin binds to both albumin and α- and β-globulins; both riboflavin and the coenzymes also bind to immunoglobulins.
Uptake into tissues is by passive carrier-mediated transport of free riboflavin, followed by metabolic trapping by phosphorylation to riboflavin phosphate, and onward metabolism to FAD.
Riboflavin phosphate and FAD that are not bound to proteins are rapidly hydrolyzed to riboflavin, which diffuses out of tissues into the bloodstream. Ribofla-vin and riboflavin phosphate that are not bound to plasma proteins are filtered at the glomerulus; renal tubular resorption is saturated at normal plasma con-centrations. There is also active tubular secretion of the vitamin; urinary excretion of riboflavin after moderately high doses can be two- to threefold greater than the glomerular filtration rate.
Under normal conditions about 25% of the urinary excretion of riboflavin is as the unchanged vitamin, with a small amount as glycosides of riboflavin and its metabolites.
Riboflavin balance
There is no significant storage of riboflavin; apart from the limitation on absorption, any surplus intake is excreted rapidly, so that once metabolic requirements have been met urinary excretion of riboflavin and its metabolites reflects intake until intestinal absorption is saturated. In depleted animals, the maximum growth response is achieved with intakes that give about 75% saturation of tissues, and the intake to achieve tissue saturation is that at which there is quantitative excretion of the vitamin.
There is very efficient conservation of riboflavin in deficiency, and almost the only loss from tissues will be the small amount that is covalently bound to enzymes and cannot be salvaged for reuse. There is only a fourfold difference between the minimum con-centration of flavins in the liver in deficiency and the level at which saturation occurs. In the central nervous system there is only a 35% difference between defi-ciency and saturation.
Metabolic functions of the flavin coenzymes
The metabolic function of the flavin coenzymes is as electron carriers in a wide variety of oxidation and reduction reactions central to all metabolic processes, including the mitochondrial electron transport chain, and key enzymes in fatty acid and amino acid oxida-tion, and the citric acid cycle. The flavin coenzymes remain bound to the enzyme throughout the catalytic cycle. The majority of flavoproteins have FAD as the prosthetic group rather than riboflavin phosphate; some have both flavin coenzymes and some have other prosthetic groups as well.
Flavins can undergo a one-electron reduction to the semiquinone radical or a two-electron reduction to dihydroflavin. In some enzymes formation of dihy-droflavin occurs by two single-electron steps, with intermediate formation of the semiquinone radical. Dihydroflavin can be oxidized by reaction with a substrate, NAD(P)+, or cytochromes in a variety of dehydrogenases, or can react with molecular oxygen in oxygenases and mixed function oxidases (hydroxylases).
Flavins and oxidative stress
Reoxidation of the reduced flavin in oxygenases and mixed function oxidases proceeds by way of formation of the flavin radical and flavin hydroperoxide, with the intermediate generation of superoxide and perhy-droxyl radicals and hydrogen peroxide. Because of this, flavin oxidases make a significant contribution to the total oxidant stress of the body. Overall, some 3–5% of the daily consumption of about 30 mol of oxygen by an adult is converted to singlet oxygen, hydrogen peroxide, and superoxide, perhydroxyl, and hydroxyl radicals, rather than undergoing complete reduction to water in the electron transport chain. There is thus a total production of some 1.5 mol of reactive oxygen species daily, potentially capable of causing damage to membrane lipids, proteins, and nucleic acids.
Riboflavin deficiency
Although riboflavin is involved in all areas of metabo-lism, and deficiency is widespread on a global scale, deficiency is not fatal. There seem to be two reasons for this. One is that, although deficiency is common, the vitamin is widespread in foods and most diets will provide minimally adequate amounts to permit main-tenance of central metabolic pathways. The second, more important, reason is that in deficiency there is extremely efficient reutilization of the riboflavin that is released by the turnover of flavoproteins, so that only a very small amount is metabolized or excreted.
Riboflavin deficiency is characterized by lesions of the margin of the lips (cheilosis) and corners of the mouth (angular stomatitis), a painful desquamation of the tongue, so that it is red, dry, and atrophic (magenta tongue), and a seborrheic dermatitis, with filiform excrescences, affecting especially the nasola-bial folds, eyelids, and ears.
There may also be conjunctivitis with vasculariza-tion of the cornea and opacity of the lens. This last is the only lesion of ariboflavinosis for which the bio-chemical basis is known: glutathione is important in maintaining the normal clarity of crystallin in the lens, and glutathione reductase is a flavoprotein that is particularly sensitive to riboflavin depletion.
The main metabolic effect of riboflavin deficiency is on lipid metabolism. Riboflavin-deficient animals have a lower metabolic rate than controls and require a 15–20% higher food intake to maintain body weight. Feeding a high-fat diet leads to more marked impair-ment of growth and a higher requirement for ribofla-vin to restore growth.
Resistance to malaria in riboflavin deficiency
Several studies have noted that in areas where malaria is endemic, riboflavin-deficient subjects are relatively resistant and have a lower parasite burden than ade-quately nourished subjects. The biochemical basis of this resistance to malaria in riboflavin deficiency is not known, but two possible mechanisms have been proposed.
● The malarial parasites may have a particularly high requirement for riboflavin. Some flavin analogues have antimalarial action.
● As a result of impaired antioxidant activity in erythrocytes, there may be increased fragility of erythrocyte membranes or reduced membrane flu-idity. As in sickle cell trait, which also protects against malaria, this may result in exposure of the parasites to the host’s immune system at a vulner-able stage in their development, resulting in the production of protective antibodies.
Riboflavin requirements
Estimates of riboflavin requirements are based on depletion/repletion studies to determine the minimum intake at which there is significant excretion of the vitamin. In deficiency there is virtually no excretion of the vitamin; as requirements are met, so any excess is excreted in the urine. On this basis the minimum adult requirement for riboflavin is 0.5–0.8 mg/day.
At intakes of 1.1–1.6 mg/day urinary excretion rises sharply, suggesting that tissue reserves are saturated.
A more generous estimate of requirements, and the basis of reference intakes, is the level of intake at which there is normalization of the activity of the red cell enzyme glutathione reductase; the activity of this flavoprotein is especially sensitive to riboflavin nutri-tional status. Normal values of the activation coeffi-cient are seen in subjects whose habitual intake of riboflavin is between 1.2 mg/day and 1.5 mg/day.
Assessment of riboflavin nutritional status
The urinary excretion of riboflavin and its metabo-lites (either basal excretion or after a test dose) can be used as an index of status. However, riboflavin excre-tion is only correlated with intake in subjects who are in nitrogen balance. In subjects in negative nitrogen balance there may be more urinary excretion than would be expected, as a result of the catabolism of tissue flavoproteins, and loss of their prosthetic groups. Higher intakes of protein than are required to maintain nitrogen balance do not affect the require-ment for riboflavin or indices of riboflavin nutri-tional status.
Glutathione reductase is especially sensitive to riboflavin depletion. The activity of the enzyme in erythrocytes can therefore be used as an index of riboflavin status. Interpretation of the results can be complicated by anemia, and it is more usual to use the activation of erythrocyte glutathione reductase (EGR) by FAD added in vitro. An activation coeffi-cient of 1.0–1.4 reflects adequate nutritional status, whereas >1.7 indicates deficiency.
Interactions with drugs and other nutrients
The phenothiazines such as chlorpromazine, used in the treatment of schizophrenia, and the tricyclic anti-depressant drugs such as imipramine, are structural analogues of riboflavin, and inhibit flavokinase. In experimental animals, administration of these drugs at doses equivalent to those used clinically results in an increase in the EGR activation coefficient and increased urinary excretion of riboflavin, with reduced tissue concentrations of riboflavin phosphate and FAD, despite feeding diets providing more riboflavin than is needed to meet requirements. Although there is no evidence that patients treated with these drugs for a prolonged period develop clinical signs of ribo-flavin deficiency, long-term use of chlorpromazine is associated with a reduction in metabolic rate.
Riboflavin deficiency is sometimes associated with hypochromic anemia as a result of impaired iron absorption. A greater proportion of a test dose of iron is retained in the intestinal mucosal cells bound to ferritin, and hence lost in the feces, rather than being absorbed, because the mobilization of iron bound to ferritin in mucosal cells for transfer to transferrin requires oxidation by a flavin-dependent enzyme.
Riboflavin depletion decreases the oxidation of dietary vitamin B6 to pyridoxal; pyridoxine oxidase is a flavoprotein and is very sensitive to riboflavin deple-tion. It is not clear to what extent there is functional vitamin B6 deficiency in riboflavin deficiency. This is partly because vitamin B6 nutritional status is gener-ally assessed by the metabolism of a test dose of tryp-tophan, and kynurenine hydroxylase in the tryptophan oxidative pathway is a flavoprotein; riboflavin defi-ciency can therefore disturb tryptophan metabolism quite separately from its effects on vitamin B6 nutri-tional status.
The disturbance of tryptophan metabolism in riboflavin deficiency, due to impairment of kynuren-ine hydroxylase, can also result in reduced synthesis of NAD from tryptophan, and may therefore be a factor in the etiology of pellagra.
Related Topics