Chapter: Biology of Disease: Disorders of the Blood
Sickle Cell Anemia - Hemoglobinopathies
SICKLE CELL ANEMIA
Sickle cell anemia was first described in a black patient in the
USA in 1904, a time when little was known of the structures of proteins. The
patient presented with severe pain and a microscopic examination of a blood
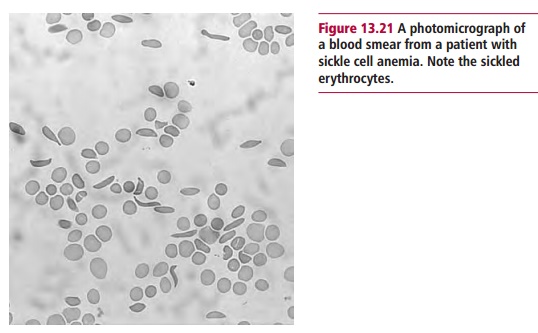
sample showed sickle shaped erythrocytes (Figure
13.21). The gene for sickle cell Hb (HbS) differs from that for HbA by a
single point mutation in the β-globin gene at the codon
responsible for the amino acid residue at position 6. This substitutes a
thymine for an adenine base. Given that one β-globin gene is inherited from
each parent, the condition may be homozygous (HbSHbS) or
heterozygous (HbAHbS).
The mutation means that an acidic, hydrophilic glutamate residue
is replaced by a hydrophobic valine. The presence of the valine residue means
that the Hb molecule is a little more hydrophobic or ‘sticky’ in two places on
its surface because there are two β-chains present. The sticky
patches are more exposed in the deoxygenated state when the conformation of HbS
changes as the molecule releases its oxygen in the tissues. The HbS molecules
therefore aggregate forming stiff fibrils that cause the sickling of the
erythrocytes although, even after years of study, it is still not completely
understood how these changes occur. The deformed erythrocytes are less flexible
than normal ones and cannot squeeze through the capillaries in the tissues and
block them. This leads to hemostasis,
anoxia and severe pain and, because the sickling occurs as a result of changes
occurring in deoxyhemoglobin, the effects are exacerbated and more cells become
sickle-shaped. The life of a typical erythrocyte is reduced from 120 to about
10 12 days in sickle cell patients: the abnormal cells
are destroyed in the spleen and consequently anemia ensues.
At low concentrations, HbS shows a normal oxygen binding curve,
but at high concentrations, as would occur in the erythrocytes of a homozygote
for sickle cell anemia, the oxygen affinity is decreased. Again, the reasons
for this are not fully understood, although the resulting shift in the oxygen
dissociation curve to the right means a greater proportion of the oxygen is released
and ameliorates the effects of the anemia. Indeed, the amino acid substitution
that causes the condition does not affect the structure of the oxygen binding
site or the ability of the molecule to bind and carry oxygen.
Patients who are homozygous for sickle cell anemia present with
crises of intense pain that can occur anywhere in the body caused by blockage
of capillaries. Crises tend to occur when the circulation is slow or when there
is hypoxia; about 15 s of low oxygen tension are required to produce sickling
so when the circulation is reasonably rapid there is insufficient time for this
to happen. Clinical complications of sickle cell disease are highly variable,
and the clinical consequences may include megaloblastic erythropoiesis,
aplastic crises, stroke, bone pain crises, proneness to infection, especially
by Pneumococcus, Salmonella and Haemophilus
due to hyposplenism, and acute chest syndrome. Acute chest syndrome is a common
form of crisis in children with sickle cell disease and is sometimes fatal. It
occurs in about 40% of all people with sickle cell disease. It is characterized
by severe chest pain and difficulty in breathing. It is probably caused either
by a chest infection or by blocked pulmonary capillaries resulting from a blood
clot. In developed countries the mortality in sickle cell disease is relatively
low but this is not the case in developing countries. In general, there seems
to be an approximate 10% mortality in the first few years of life but, again,
this depends on the treatment available, namely whether the infant has
‘Western-style’ medical care. The probability of surviving to 29 years is about
84% but there are few data on longevity. Infections seem to be the commonest
cause of death at all ages.
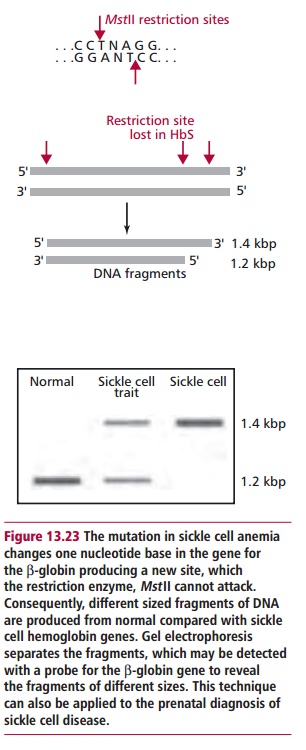
A diagnosis of sickle cell anemia may now be carried out on the
DNA (Figure13.23) of the embryo
obtained by chorionic villus sampling or amniocentesis.The parents may then
make an informed decision whether or not to continue with the pregnancy.
Treatment includes analgesia for the pain during crises and antibiotics and
vaccination against the likely life-threatening infections. The pain is often
so acute as to require morphine. Sometimes inhalation of nitric oxide can help
by producing a vasodilation but this treatment is only dealing with the
symptoms. Blood transfusions are also possible but they can lead to iron
overload, as well as other complications, as the transfused erythrocytes are
removed from the circulation. Chelating agents such as desferrioxamine may be
used. In was observed that the severity of sickle cell disease in some
populations was reduced by the presence of high concentrations of HbF. Fetal Hb
is almost as good as HbA in transporting oxygen and, of course, does not
sickle. Hydroxyurea and butyrate are used as therapeutic agents to try to
induce higher levels of HbF in sickle cell patients. Hydroxyurea is thought to
kill selectively precursor cells in the bone marrow whilst sparing the
erythroblasts that produce HbF. However, this compound is an antineoplastic agent
and its long-term effects are unknown. Butyrate seems to activate transcription
of the γ-globin gene so that HbF is produced in the adult.
Both agents have met with reasonable success in treating sickle cell patients
and, in some cases, may be used synergistically to increase HbF up to 20% with
a marked clinical improvement.
The mutation that causes HbS production is not the only one that
leads to sickling of erythrocytes but the many other variants are rather rare.
The second commonest of these in black Americans occurs in HbC, in which a
lysine residue replaces glutamate at position 6 in the β β chain. Hemoglobin C is rather
insoluble and crystals of it can sometimes be seen in peripheral blood smears.
Heterozygotes for HbC are asymptomatic but homozygotes have a mild hemolytic
anemia.
As a result of the coincident distribution of the genes for HbS
and HbC, heterozygotes for both Hbs are not uncommon producing HbSC disease.
This is milder than true sickle cell disease but patients can show practically
all the same complications. Furthermore, it is symptomatic in the heterozygous
state. There can also be co-inheritance of the sickle cell and thalassemia
genes, which generates a wide spectrum of clinical symptoms whose severity
depends on the type of thalassemia mutation (see below).
Related Topics