Chapter: Biology of Disease: Disorders of the Blood
Hemoglobinopathies
HEMOGLOBINOPATHIES
Hemoglobinopathies are clinical conditions that result
from mutations thatchange the sequences of bases in DNA of the genes for
globins If the bases in the DNA are
changed even by a single one, then a modified protein may be produced (or no
protein at all). The consequences can be negligible, severe or fatal. Mutations
are inherited and, if the disease is not fatal, then the disease symptoms will
be inherited too. The severity of the disease may depend on whether one or both
copies of the gene in question carry the mutation, in other words, whether the
individual is homozygous or a heterozygote. The mutations involved in
hemoglobinopathies include point mutations, the largest group, that substitute
one amino acid residue for another, insertions or deletion of one or more
residues, drastic changes caused by frameshift mutations (Margin Note 13.6) and alterations in the lengths of the polypeptide
chains by mutations that produce or destroy stop codons.
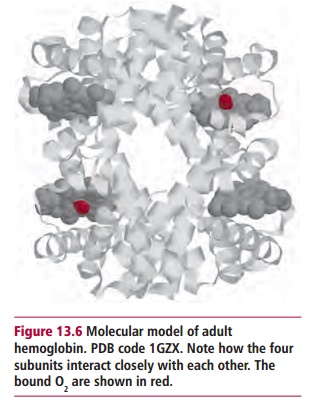
In normal adult humans, there are two α - and one β-globin genes, coding for
polypeptides of 141 and 146 amino acid residues respectively, which go to form
HbA, α 2 β 2. In a diploid cell there are
actually four α and two β genes. Each of these genes has two introns (Margin Note 13.7). The α genes are located on chromosome 16 and the β genes on chromosome 11. If there is a mutation, it
may have been inherited from one or both parents giving a heterozygous or
homozygous condition respectively. A mutation in an α gene tends to have less serious consequences than
one in a β gene because there may still be
nonmutated copies of the α gene present. Nevertheless, even
small changes in the structure of the Hb protein can sometimes result in
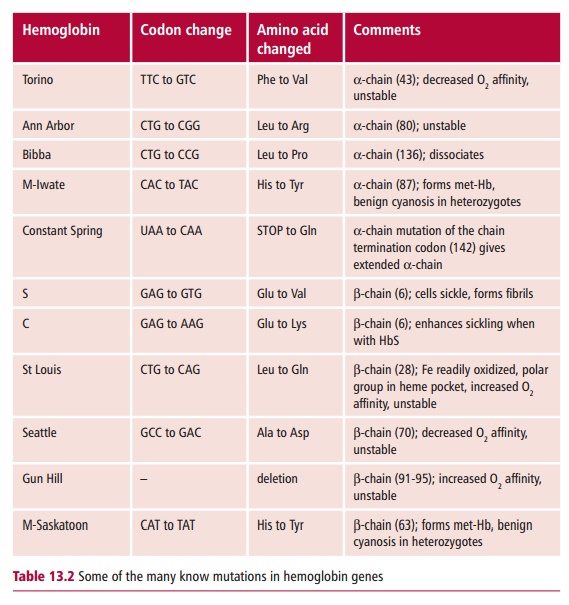
disastrous clinical effects. Over 750 Hb mutations are known. They usually only
affect one type of subunit because there are separate genes for the α - and the β-globins (Table13.2).
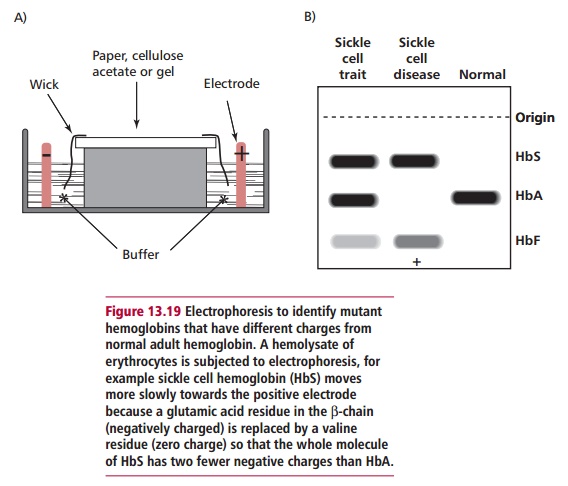
Originally, many of the different mutant Hbs were identified by
their mobilities in electrophoresis (Figure
13.19) and peptide mapping but now, of course, the DNA can be analyzed
directly. A major technical advance has been the ability to make DNA probes
that are specific for α -or β-chains. This means it is possible to identify which mRNAs are
being produced and identify any mutations present. Thus the different clinical variants can be understood at
the molecular level. For example, in
so-called ‘hemoglobin H disease’ it has been shown that there is only one of
the four possible α -globin genes present and
functioning, so that only 25% of the normal amount of α -chain mRNA is produced. The mutation causing this
situation is a deletion not a point mutation.
The majority of mutations are harmless and therefore do not produce a hemoglobinopathy because they do not cause disease. For example, mutations distant from the heme binding cleft, or from the regions of
subunit contact may have little effect on the properties of the Hb. However,
mutations may change the shape of the globin subunit(s), the binding of the
heme groups or even prevent globin synthesis, all with severe clinical
consequences. To function properly, the four subunits in the Hb molecule must
fit together tightly but still produce a molecule that is flexible. The regions
of contact have been conserved in evolution and are essential for normal
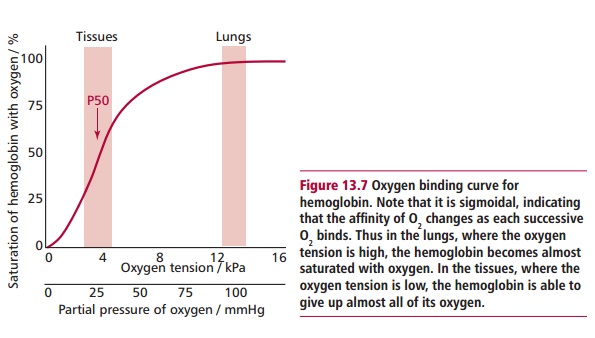
functions, such as the cooperative binding of O2 (Figure 13.7). Thus mutations can upset
the delicate balance of interactions between the amino acid side chains with
several consequences. The molecule may dissociate upon deoxygenation and, in
some cases, the monomers may precipitate in the erythrocytes reducing O2
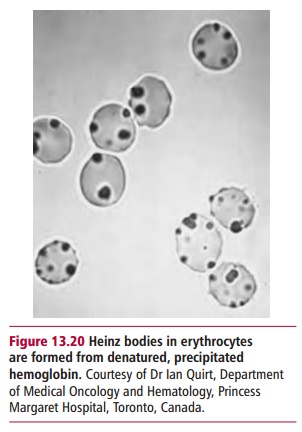
affinity. Microscopically, the denatured and precipitated Hb can be seen as
Heinz bodies (and Figure 13.20). A deletion of one or more
amino acid residues or substitution mutations can produce this effect, as in Hb
Leiden and Hb Philly respectively. There may also be cell membrane damage, with
intravascular hemolysis, anemia, reticulocytosis and splenomegaly as
consequences. In other cases, a small change in the regions that bind the heme
groups may make the pockets slightly less hydrophobic so that it does not bind
appropriately, and again, the denatured Hb can precipitate to form Heinz
bodies. Thus only two of the four subunits may have heme groups. In other
cases, the change in the pocket allows the iron to become oxidized to the Fe3+(III)
state (methemoglobin), which will not bind O2. The resulting
condition is referred to as methemoglobinemia and patients become cyanosed
because they lack oxygen.
Related Topics